![[GRAPH OF SURVIVAL AGAINST AGE]](lifespan.gif)
Note: This monograph does NOT have a terminal copyright date — development is ongoing
Aging is a syndrome of changes that are deleterious, progressive, universal and thus far irreversible. Aging damage occurs to molecules (DNA, proteins, lipids), to cells and to organs. Diseases of old age (diseases which increase in frequency with age, such as arthritis, osteoporosis, heart disease, cancer, Alzheimer's Disease, etc.) are often distinguished from aging per se. But even if the aging process is distinct from the diseases of aging, it is nonetheless true that the damage associated with the aging process increases the probability that diseases of old age will occur.
Some gerontologists prefer to use the word senescence because "aging" implies that the passage of time necessarily results in deterioration (biological entropy) — which is certainly not true during the early, developmental, time of life (before the age of 10 or 12 in humans). I will retain the word "aging" because I believe the association between aging & deterioration is universal as adult years progress and because the distinction between aging & development is very strongly established in conventional language. Also, shorter words make for slightly faster reading.
One can catalog changes that typically occur with age. For people of developed countries age changes include: A loss of hearing ability, particularly for higher frequencies. There is a decline in the ability to taste salt&bitter (sweet&sour are much less affected). There is a reduction of the thymus gland to 5−10% of its original mass by age 50. Levels of antibodies increase with aging. One third of men and half of women over 65 report some form of arthritis. About half of those aged 65 have lost all teeth. The elderly require twice as much insulin to achieve the glucose uptake of the young. There is reduced sensitivity to growth factors & hormones due to fewer receptors and dysfunctional post-receptor pathways. The temperature needed to separate DNA strands increases with age. Weight declines after age 55 due to loss of lean tissue, water and bone (cell mass at age 70 is 36% of what it is at age 25). Body fat increases to age 60. Muscle strength for men declines 30−40% from age 30 to age 80. Reaction time declines 20% from age 20 to 60. Elderly people tend to sleep more lightly, more frequently and for shorter periods — with a reduction in rapid eye-movement (REM) sleep. Neurogenesis in the hippocampus declines with age. Degree of saturation of fats drops by 26% in the brains of old animals. Presbyopia (reduced ability to focus on close-up objects) occurs in 42% of people aged 52−64, 73% of those 65−74 and 92% of those over age 75. Most people over age 75 have cataracts. About half of those over 85 are disabled (defined as the inability to use public transportation). Over 75% of people over 85 have 3−9 pathological conditions, and the cause of death for these people is frequently unknown.
Aging changes are frequently associated with an increase in likelihood of mortality, but this is not necessarily the case. For example, graying of hair is a symptom of aging, but graying does not increase likelihood of mortality. Aging changes which are not associated with a specific disease, but which are associated with a generalized increase in mortality would qualify as biomarkers of aging — and would distinguish biological age from chronological age. Biomarkers would be better predictors of the increased likelihood of mortality (independent of specific disease) than the passage of time (chronological age). Cross-linking of collagen, insulin resistance and lung expiration capacity have been proposed as candidates but, as yet, no biomarkers of aging have been validated and universally accepted.
Many scientists have wondered whether a single cause (probably cellular
or hormonal) lies behind all aging phenomena — or whether aging is inherently
multi-faceted. Differences in lifespan between species raise critical
questions, in this regard. Why is a rodent old at 3 years, a horse old at
35 years and a human old at 80 years? Aren't the cells much the same? Why
is it that at age 3 about 30% of rodents have had cancer, whereas at age
85, about 30% of humans have had cancer? Some species (such as lobsters,
alligators and sharks) show few signs of aging. Cancer cells, stem
cells and human germ cells seem "immortal" when compared to other cells.
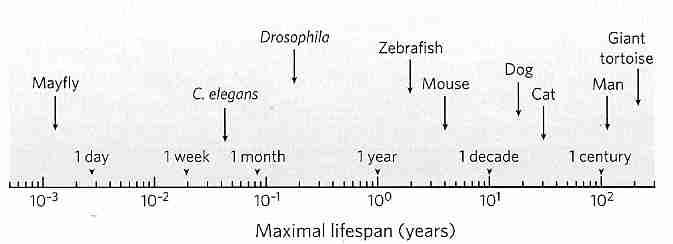
When discussing aging it is important to distinguish two points on survival curves. Mean lifespan (average lifespan) corresponds to the age at which the horizontal line for 50% survival intersects the survival curve. Maximum lifespan corresponds to the age at which the survival curves touch the age-axis (0% survival) — and this represents the age at which the oldest known member of the species has died. (In animal studies, maximum lifespan is typically taken to be the mean lifespan of the most long-lived 10%.) Curve A as shown is a pure exponential decay curve. Curve B corresponds to the survival of small animals, such as mice or squirrels in a natural environment. Human survival was still close to curve B in ancient Rome when average lifespan was 22 years, but by the mid−1800s the typical North American lived to be 40 — more like curve C. Today, people in the most developed countries have an average lifespan of about 80 — resembling curve D. Reduction of infant mortality has accounted for most of the increased longevity, but since the 1960s mortality rates among those over 80 years has been decreasing by about 1.5% per year. Maximum lifespan for humans, however, has remained about 115−120 all through known history. The longest documented human lifespan has been for Frenchwoman Jean Calment who lived 122.3 years.
Curing specific diseases such as heart disease or cancer can do no more than further "square" the survival curve (toward curve E), with no effect on maximum lifespan. Curing cancer would add about 2 years to human life, whereas eliminating heart disease would add 3 or 4 years. Mean lifespan varies with susceptibility to disease, accident & homicide/suicide, whereas maximum lifespan is determined by "rate of aging". In aging research, maximum lifespan is regarded as a proxy for aging. Chemicals, calorie restriction with adequate nutrition, or other interventions which increase maximum lifespan are said to have slowed the aging process.
If human beings were free of disease & senescence the only causes of death would be accident, suicide & homicide. Under such conditions it is estimated that from a population of one billion, a 12-year-old would have a median lifespan of 1,200 years and a maximum lifespan of 25,000 years.
In 1825 an English actuary named Benjamin Gompertz discovered that likelihood of dying increases exponentially with age after maturity — an empirical observation that has stood the test of time. A 35-year-old is twice as likely to die as a 25-year-old and a 25-year-old is twice as likely to die as a 15-year-old. The exponential increase does not continue past age 80 and death rate may even decline after age 110 [SCIENCE 280:855-860 (1998)]. (Medflies — Mediterranean fruit flies — show a plateau of linear rather than exponential death rate when 20-25% of the population remains). Similarly, the risk of getting Alzheimer's Disease doubles every 5 years past the age of 60 — probably plateauing after age 90 (when over half the population is already demented). Cancer rate increases exponentially with age, but also seems to plateau in the very elderly. One explanation might be that subsets of the population that are considerably more hardy due to genetics or behavior may remain after the more heterogenous majority have died. Another explanation suggests the complete elimination of the forces of natural selection at the oldest ages — which causes subsequent survival to be completely the result of genetic "random drift" [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA) 93:15249-15253 (1996)]. Causes of death in middle-age tend to be due to diseases affecting high-risk individuals (cancer, diabetes, hypertension, etc.), whereas the elderly are more vulnerable to multiple pathologies due to vulnerability of aging organs & tissues [JOURNALS OF GERONTOLOGY 58A(6):B495-B507 (2003)].
Attempts to classify theories of aging have led to the two major classifications programmed aging and wear&tear aging. Programmed aging would be aging due to something inside an organism's control mechanisms that forces elderliness & deterioration — similar to the way genes program other life-stages such as cell differentiation during embryological development or sexual maturation at adolescence. By contrast aging due to wear&tear is not the result of any specific controlling program, but is the effect of the sum effect of many kinds of environmental assaults — ie, damage due to radiation, chemical toxins, metal ions, free-radicals, hydrolysis, glycation, disulfide-bond cross-linking, etc. Such damage can affect genes, proteins, cell membranes, enzyme function, blood vessels, etc.
When Pacific salmon have lived in the ocean for 2 or 3 years, they make an arduous upstream journey against a raging riverswim until they find a place suitable for spawning. After spawning, the adrenal gland releases massive amounts of corticosteroids — leading to rapid deterioration. It would be costly for the species to have salmon that could live another year and repeat the journey — or compete with the offspring for food. Although this process is obviously "programmed", it is inaccurate to describe it as "aging". Programmed death, rather than programmed aging, is a common phenomenon among animals that reproduce only once.
Grazing animals show wear-and-tear to their teeth to the point where they can no longer eat, and they die of starvation. Again, it stretches the point to say the teeth are aging. The teeth of rabbits (like human fingernails) continue to grow as wearing occurs — and in this sense are "programmed" to compensate for "wear&tear". Why don't grazing animals have teeth that continue to grow? Human beings can replace tissue, capillaries and bone in wound-healing, yet cannot regrow a severed limb the way a salamander can. Why isn't human DNA "programmed" to re-grow kidney or liver tissue as it ages? Planarians (flatworms) have a pool of stem cells which can replace any of their fully differentiated cells. Programming that compensates for wear & tear should be distinguished from programming that causes deterioration.
Russell Wallace, who with Charles Darwin discovered natural selection, speculated that longevity much beyond the age of procreation would be a disadvantage for a species. Parents would threaten their children by competition for resources. This would imply an evolutionary advantage to genetically programmed aging. The programmed self-destruction with corticosteroids by Pacific salmon after spawning — and whose decaying bodies provide nutrient for their offspring — may be severe example indicating the possiblity of programmed senescence. But as biologist Peter Medawar noted, there is circular reasoning in claiming that senescence evolved so that non-senescent individuals could more readily survive. If there were no senescent, poorly-reproducing individuals, there would be no need for replacement.
If aging were the product of evolutionary forces, aging could reasonably be expected to result from programming. But since most animals in the wild die of accident, attack or disease it seems questionable that evolutionary forces determine aging. Robins in the wild, for example, have an estimated 12-year maximum lifespan and a 40% chance of surviving any given year. With a (0.4)12 — or 1 in 60,000 — chance that a robin can avoid accident, attack or disease for 12 years, there would seem to be little opportunity for natural selection to play a role in the evolution of senescence. Against this argument is evidence that early stages of senescence reduce the ability of an animal to survive — thereby causing earlier selection against older animals.
An alternative to the view that senescence is the product of evolution compares genetic programming to the engineering of a fly-by satellite designed to gather data about a planet. The engineering is focused on ensuring that the satellite reaches its destination and performs its data gathering/transmission when passing the planet. Beyond the planet it is a matter of indifference to the engineers how long the satellite continues to function — random decay occurs. Applying the analogy, the satellite passing the planet is like an organism passing its reproductive period. Once the objectives of reproduction & parenting have been achieved the organism decays by random malfunction.
|
The vast range of maximum lifespan differences between species provides convincing evidence that longevity is genetically influenced. An elephant lives about 10−20 times longer than a mouse, yet both animals have roughly the same number of lifetime heartbeats — the elephant at 30 per minute and the mouse at 300 per minute. Both species take about 200 million breaths in a lifetime. And both species have a metabolic potential (total kilocalories used per gram of body weight per lifetime) of about 200 kcal. This figure is much the same for other mammals, but humans are exceptional with a metabolic potential of 800 kcal. Brains use more energy than any other human organ. (Basal metabolic rate for humans is about 80 watts = 70 Calories per hour.) Birds have a metabolic potential of 1,000 to 1,500 kcal.
Gerontologists who compare the longevity of species explain this discrepancy by saying that while body weight correlates well with longevity, there is a better correlation with brain weight for primates. For other species brain size may be more related to motor function than to cognitive capacity.
Flight, like brain weight, also confers a longevity advantage. Finches & robins live about 3 times as long as rodents the same size. Flying squirrels live twice as long as their close relatives the chipmunks. Parrots have a maximum lifespan in excess of 90 years. The Andean condor may be the most long-lived of any bird, but its maximum lifespan has not been confirmed.
Gross attributes of species typically associated with greater longevity are: large size, ability to fly, brainy, a spiny or shelled encasement, and cold-blooded. All but the last attribute reduce vulnerability to predators. Porcupines are the longest-lived rodents. Naked mole rats, by living underground, are also safer from predators and live significantly longer than similarly-sized rats. All adaptations that afford protection from predators and other hazards justify greater developmental resources to build a more durable animal with a longer maximum lifespan.
Opossums evolving on an island free of predators have been shown to have substantially longer lifespans and smaller litters than opossums living on the nearby mainland [JOURNAL OF ZOOLOGY; 229:695-708 (1993)]. Where competition between individuals of a species for mates & resources is more important than survival against predators and other hazards, evolution causes more investment in making a more hardy & durable animal — which includes having fewer offspring on each birthing (but more total offspring over the lifetime) — with each offspring receiving more care and resources. Gene survival can be better promoted (up to a point) by extending lifespan and reproductive period of reproductively successful adults than by creating many more offspring, a signficant number of whom will not survive to become reproductive adults.
Large size also confers protection against predators and confers an improved ability to escape dangerous environments. Metabolic rate decreases proportionally with increases in body size, which allows larger animals to survive longer when food & water are scarce. [For a sphere, surface area S = 4πr2 and volume V = (4/3)πr3, which means that S/V varies inversely with r (radius). Because heat is generated in the volume and dissipates in the surface area, relative dissipation decreases with an increase in radius because of the decrease in S/V.] Large animals are better able to withstand extreme temperatures because of greater body mass. Large animals and birds are more easily able to travel long distances to find food or less harsh environments.
Cold-blooded animals needn't expend energy to maintain body temperature and therefore generate fewer free-radicals. Also, the rate of chemical reactions more than doubles for each 10ºC increase in temperature. Cold-blooded animals may use one-tenth as much energy as warm-blooded animals of the same body weight. The alligator, Galapagose tortoise and lake sturgeon combine large size with cold-bloodedness. Turtles live longer than other reptiles because of the shell which protects against predators. With the combination of hard shell, large size and cold-bloodedness, it is not surprising that the Galagose turtle is probably the most long-lived vertebrate. Hard shell, cold-bloodedness and the ability to reduce metabolic rate allow some bivalves to live nearly four centuries [GERONTOLOGY; Philipp,EER; 56(1):55-65 (2010)].
A short-lived organism would waste metabolic energy by over-investing in anti-oxidant or DNA-repair enzymes when the energy could be spent on rapid growth and reproduction. When a species has fewer predators, evolution invests fewer resources into speedy reproduction and more genetic resources (DNA repair, etc.) into a longer reproductive period (longer life). In the case of birds, the mitochondrial membranes contain more unsaturated fat making them less vulnerable to lipid peroxidation. And the protein complexes of the respiratory chain of mitochondria generate fewer free radicals in birds than in mammals. It is conceivable that an animal with well-engineered cells could live many centuries. Human germ cells have arguably lived for millions of years through an investment in DNA-repair enzymes, antioxidant enzymes and telomerase.
Evolutionary biologists are able to use artificial selection in the laboratory experimentally (rather than passively studying natural selection in the wild) to seek the evolutionary determinates of longevity. Michael Rose at the University of California has shown that Drosophila (fruit-flies) bred for 15 generations by disposing of eggs laid early in life and only using eggs that were laid toward the end of reproductive life achieved maximum lifespans 30% greater than that of controls. The long-lived strains had increased levels of SOD, CAT and xanthine dehydrogenase as well as increased levels of heat shock proteins conferring stress resistance [JOURNALS OF GERONTOLOGY 55A(11):B552-B559 (2000)]. Hsp22 heat shock protein expression was 2−10 times greater in the long-lived strains as compared to controls. Transgenic Drosophila (ie, fruit flies with artificially altered genes) with extra copies of hsp70 genes live nearly 8% longer than controls following heat treatment [NATURE; Tatar,M; 390:30 (1997)].
Dr. Rose has also observed the experimental increase in mortality associated with aging ceases late in life [PHYSIOLOGICAL AND BIOCHEMICAL ZOOLOGY; Rose;MR; 78(6):869-878 (2005)]. Although mortality rates remain very high in late-life, they plateau. Studies of inbred Drosophila indicate that the plateauing cannot be due to genetic variation. From his evolutionary biology perspective Dr. Rose associates this phenomenon with a late-life end of the force of natural selection. This would imply that senescence is genetically programmed and that studying the genetics of the plateau could be the key to understanding the genetics of longevity.
In nearly every culture on earth women outlive men — significantly
so in the oldest years. But the men who do survive to become
elderly are hardier than the women. A US National Institute of Aging study
showed that 44% of men over age 80 are "robust and independent"
compared with only 28% of women. And the percentage of surviving males
increases from 15% at age 100 to 40% at age 105 in the United
States.
![[Graph of
Fertility Decline]](fertile2.gif)
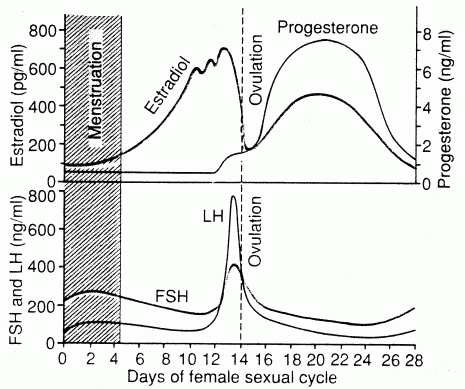
If aging has been programmed by evolutionary forces, sex could be a very important contributor to the program. The reproductive organs of the human female exhibits a rate of aging that is among the most rapid of body systems. The complete shutdown of female fertility at menopause may be of value in preventing the birth of deformed children or death in childbirth of a mother who has several dependent children. For a species with a lengthy parenting period, it makes sense for fertility to cease long before the debilities of advancing age begin. Gonadotropin hormones from the pituitary gland are controlled by gonadotropin-releasing hormone, a 10-amino-acid peptide originating in neurons located in the arcuate nucleus of the hypothalamus. The two gonadotropin hormones (FSH & LH) are the same for females as for males, although their function is very different. Simplistically, FSH stimulates egg production in females & sperm production in males, whereas LH stimulates estrogen production in females & testosterone production in males.
In fertile females FSH
(Follicle-Stimulating Hormone) accelerates the growth of 6−12 primary
follicles in the ovary each month — one of which may become a mature
ovum. The follicles secrete estrogens, the most powerful of which
is estradiol. A sudden increase in LH (Luteinizing Hormone) usually
triggers ovulation (follicle rupture with discharge of the ovum) and
the conversion of the follicle to the corpus luteum ("yellow body")
— which also secretes estrogen, but primarily secretes progesterone.
Progesterone stimulate the walls of the uterus to prepare it for implantation
of the fertilized ovum. If pregnancy occurs, progesterone inhibits
ovulation (by suppressing FSH & LH) and promotes uterine development
until the placenta becomes more mature. (Progesterone is so-named
because it promotes gestation, ie, the growth of offspring in the
womb).
|
Aside from their role in the monthly cycle, estrogens are responsible for the development and maintenance of the female sexual organs, cause the deposition of fat in the breast&buttocks (which contributes to the feminine figure) and have a potent effect on bone development.
Menopause is the event in a woman's life when her ovary literally runs out of eggs. The loss of follicles to produce estradiol causes an end to menstrual cycling and production of estrogen & progesterone by the ovary. At age 30, a woman's period is normally 28−30 days, but by age 40 her period is typically closer to 25 days and her rate of egg-loss has accelerated. Further shortening (accompanied by periods when no ovulation occurs) eventually leads to menopause at an average age of 50 (plus or minus 10 years). The menopausal woman often experiences anxiety, irritability and fatigue. Beginning before menopause most women experience "hot flashes", ie, 3 minute surges of blood to the skin of the chest, shoulders & face leading to sudden hotness & sweating. Hot flashes are associated with a pulsatile release of LH from hypothalamic neurons associated with body temperature elevation. Estrogen therapy eliminates hot flashes. The rate of loss of ovarian follicles doubles around age 35, raising the suspicion that a hypothalamic mechanism may be the ultimate cause of menopause [SCIENCE 273:67-70 (1996)].
The most serious complications of menopause are osteoporosis and a decline in cardiovascular health. The Framingham Heart Study demonstrated that between ages 35 to 65 men have 10 times the incidence of heart attack as women — probably because estrogen protects against heart disease. Estrogen elevates HDL cholesterol and reduces LDL cholesterol in the bloodstream.
After menopause, nipples decrease in size and the surrounding alveolar tissue shrinks. Erection of these tissues with external stimulation is more difficult. Vaginal contractions during orgasm is reduced to 4−5 at 0.8-second intervals from 8−12 in young adults.
The testes have been regarded as the source of maleness
at least since ancient Rome, where eunuchs & women were not
permitted to "testify" (testis is Latin for "witness"). In
the male, LH stimulates secretion of testosterone by the
interstitial cells of Leydig in the testes. FSH stimulates
spermatogenesis in the seminiferous tubules of the testes.
Testosterone promotes development of male sexual organs in the
foetus. At puberty testosterone stimulates hair growth on the
face & pubis, causes enlargement of the larynx to deepen the
voice, increases skin thickness, causes a 50% increase in muscle mass,
promotes bone growth, increases basal metabolism up to 15% and
increases red blood cell concentration.
![[Graph of Male Testosterone Decline]](testost.gif)
There is no sudden "andropause" event in males that is comparable to the menopause event of females. Instead, testosterone levels tend to decline gradually with age. This decline occurs most dramatically in those with cardiovascular disease or a predisposition to adult-onset diabetes. Although sperm count declines, fatherhood has been verified for a male as old as 94. Semen production declines in the prostate as a man ages — and the smooth muscle is replaced by overgrowing connective tissue that enlarges the prostate, blocks urine and can lead to cancer. 85% of men over age 50 have symptoms arising from benign prostatic hyperplasia — a noncancerous overgrowth of prostate tissue possibly caused by excessive expression of the anti-apoptosis protein bcl−2 [HUMAN PATHOLOGY 27:668-675 (1996)]. In some tissues testosterone must be converted to dihydrotestosterone (by the enzyme 5−α reductase) in order to act. This occurs most notably in the prostate gland, which produces semen (a mixture of sugars, protein and water). Dihydrotestosterone has also been implicated in baldness. The European drug Permixon (an extract of the saw palmetto berry) inhibits 5−α reductase, and is used to prevent prostate hypertrophy and prostate cancer. The Life Extension Foundation sells saw palmetto berry extracts as a dietary supplement for this purpose.
Testosterone has been used in elderly men for "rejuvenation" — to restore virility & muscle strength. Testosterone increases the risk of cardiovascular disease — by increasing blood pressure, by lowering HDL cholesterol and by elevating LDL cholesterol. These same dangerous side effects are also seen in athletes who attempt to use androgens or other anabolic steroids to improve athletic performance. Eunuchs reportedly live longer, although there have been no controlled clinical trials to prove this observation. Sterilization of a dog or cat (male or female) adds a couple of years to its lifespan. Any reduction in sex hormones would be expected to reduce cell proliferation and hence reduce the probability of cancer.
Male libido peaks in mid-adolescence, and does not correlate exactly with testosterone blood levels. In elderly men it may take from 10 seconds to several minutes to get an erection, in contrast to 3−5 seconds in young men. Contractions of the penile urethra during orgasm is reduced to 1−2 contractions per 0.8-seconds from 3−4 in young adults. Ejaculatory distance is reduced from 12−24 inches to 3−5 inches.
[For more about sex and aging, see Sex Hormone Replacement in Older Adults]
Aging in the female reproductive system provides the best example of programmed aging in mammals. For many other organs — particularly the heart, brain, lung and kidney — specific disease states associated with aging are of more significance than generalized deterioration. There is wide variation in the health status of specific organs among the elderly.
Skin, lungs, muscles, blood vessels and organ-function in general is adversely affected by protein cross-linking — which is increased in diabetes. Because most of those 65 years of age have at least some symptoms of subclinical diabetes and because most of the symptoms of aging are accelerated in diabetes, diabetes figures strongly when the elderly are described in terms of averages. Generalized reduction in blood flow due to atherosclerosis also has an adverse effect on most organ systems — some more than others. Both protein cross-linking and cardiovascular deterioration are strongly influenced by genetics and environmental influences (diet, smoking, etc.).
With aging there is normally an age-related decrease in insulin sensitivity as well as of resting metabolic rate per unit of fat-free mass. These changes may not occur for those who maintain high levels of aerobic exercise [JOURNAL OF APPLIED PHYSIOLOGY; Clevenger,CM; 93(6):2105-2111 (2002) and AMERICAN JOURNAL OF PHYSIOLOGY; van Pelt,RE; 281(3):E633-E639 (2001)]. A study of very long-lived persons (over age 95) did not show a decline in resting metabolic rate [THE JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM; Rizzo,MR; 90(1):409-413 (2005)], but another study of those over age 90 did show reduced metabolic rate [JOURNAL OF GERONTOLOGY; Frisard,MI; 62A(7):752-759 (2007)]. Whether survival is due to this trait or whether the trait is a feature of aging cannot be distinguished by cross-sectional studies.
The kidney provides perhaps the most striking example of individual variation in the effects of aging. On average, kidney weight declines about 15% between ages 40 and 80. The kidney's filtering capacity for the average 90-year-old is typically half what it is for the average 20-year-old. But high blood pressure and diabetes are particularly damaging to kidney function. A 20-year longitudinal study showed no change at all among elderly men who had no health problems. If this result can be extrapolated it would mean that within the human maximum lifespan there is no significant kidney deterioration in the absence of disease conditions. (For a discussion of the issue of whether dietary protein can harm kidney function, see my essay Does Excess Protein Cause Kidney Damage?).
Cardiovascular disease is the most frequent cause of death among those over age 85. The left ventricle of the heart increases in size with age (hypertropy) due to an increase in size of the heart muscle cells that must work harder to pump blood through a circulatory system that has narrower channels and reduced elasticity. Lipofuscin content of heart muscle cells increases from about 1% in the young to over 5% in the old. Arteries thicken with age such that about three-quarters of elderly people have increased blood pressure (both systolic & diastolic). But, stated conversely, about a quarter of elderly people do not have elevated blood pressure. According to the Framingham Heart Study, systolic blood pressure is a better predictor of mortality than diastolic blood pressure. Hypertension is defined as a systolic blood pressure greater than 160mm Hg. Hypertension is present in 5% of those aged 60 and nearly one quarter of those aged 75-80. While heart attacks from ischemia account for 43% of deaths for those 65−74 years of age, it accounts for only 8% of deaths for that age group in Japan (where death-rate from stroke is much higher). (For more details concerning cardiovascular disease, risk factors and prevention — see my essays Sudden Cardiovascular Death and Prevention of Cardiovascular Disease.)
|
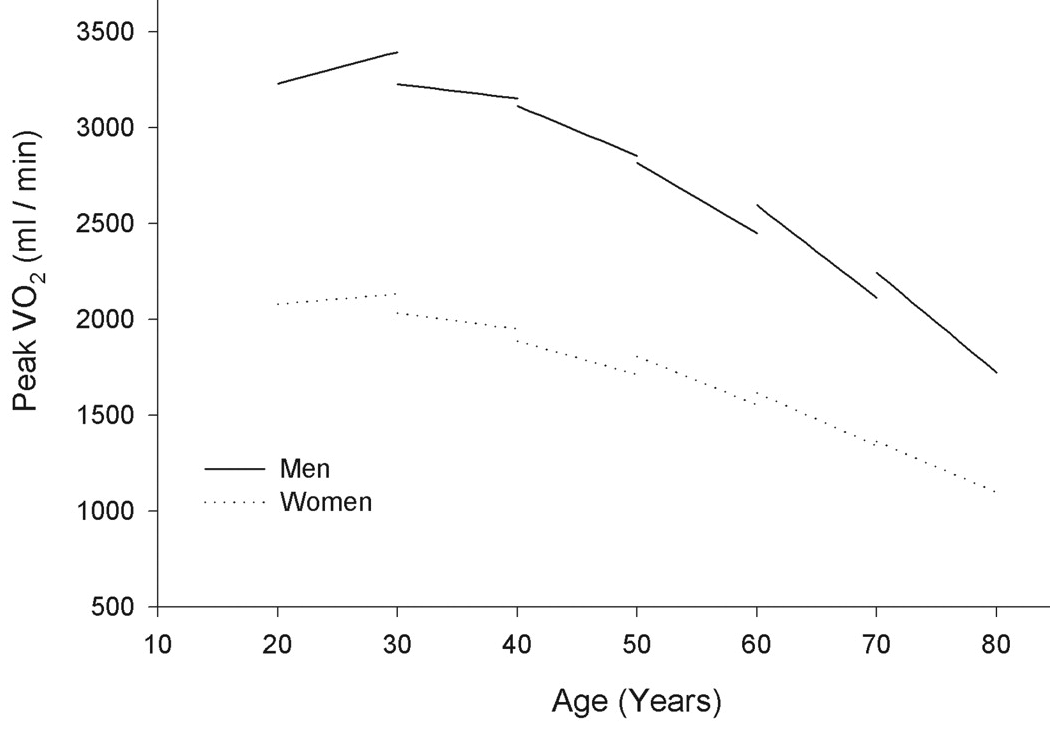
Aerobic capacity (VO2 max — liters of oxygen consumed per minute during peak exercise) declines increasingly steeply with age, and declines more steeply in men than in women. Although exercise increases aerobic capacity at any age, exercise does not prevent accelerated decline [CIRCULATION; Fleg,JL; 112(5):674-682 (2005)].
The claim that all people lose about 100,000 neurons per day has not been supported by modern research. 2% of neurons are lost, on average, between ages 20 and 90 (up to 40% of this loss in the frontal cortex). Those over age 86 show an average 10% decline in brain weight from age 20. Between age 30 and 90 brain volume declines an average of 14% in the cerebral cortex, 35% in the hippocampus and 26% in cerebral white matter. But averaging can be misleading, because the elderly include many people with considerable dementia and others with little or none. Nonetheless, a cross-sectional Magnetic Resonance Imaging (MRI) study of healthy volunteers showed age-related declines in the volume of gray matter in association area (rather than sensory areas) of the cerebral cortex, particularly in the prefrontal cortex [CEREBRAL CORTEX; Raz,N; 7(3):268-282 (1997)]. Dementias are more common among the elderly who develop cardiovascular disease. Dramatic reduction in cerebral blood flow and in brain oxygen&glucose utilization is frequently seen after the 8th decade of life. Although most dementias are due to Alzheimer's Disease, at least 20% of dementias are due to stroke(s).
Skeletal muscles are "fast-twitch" or "slow-twitch". Fast-twitch muscles ("white meat") can deliver much power over short periods through energy from anaerobic (oxygen-free) phosphagen (creatine phosphate) and glycogen/lactic-acid metabolism. Slow-twitch muscles ("dark meat") provide endurance with aerobic metabolism — using more mitochondria, more myoglobin and more capillaries per square inch. Sprinters&jumpers have more fast-twitch muscle, whereas marathoners&swimmers have more slow-twitch muscle. Posture is maintained with slow-twitch muscles. Aging results in greater loss of fast-twitch than slow-twitch muscle. Muscle fibers are replaced by fat & connective-tissue. Mitochondria die. Exercise can slow this deterioration because fast-twitch fibers atrophy due to loss of the nerves that innervate them (a loss possibly due to disuse).
Muscles in the iris of the eye atrophy, and pupil size reduces, with age — increasing the need for illumination. The lens thickens and becomes yellowed, reducing green-blue-violet discrimination. (Elderly painters use less violet & dark blue because the colors look the same.)
Collagen & elastin in tendons & ligaments become less resilient and more fragmented as a person grows older, particularly due to glycation (cross-linking of proteins by sugar). Articular cartilage becomes frayed and the synovial fluid between joints becomes "thinner". Decline in circulatory function contributes to this process. Glycation of collagen & elastin is accelerated in diabetics due to high blood sugar.
Hair graying accompanies aging regardless of gender or race. By 50 years of age approximately 50% of people have 50% gray hair [MICRON; Van Neste,D; 35(3):193-200 (2004)].
Aging of skin is commonly divided into "chronological aging" and "photoaging", with up to 80% of skin aging attributed to photoaging in non-smokers. Photoaging is due to ultraviolet (UV) light, which activates inflammatory cytokines & metalloprotein collagenases as well as inducing free radicals [ARCHIVES OF DERMATOLOGY; Fisher,GJ; 138(11):1462-1479 (2002)]. UV radiation generates singlet oxygen which both activates metalloproteinases and causes large scale deletions of mitochondrial DNA [JOURNAL OF BIOLOGICAL CHEMISTRY; Berneburg,M; 274(22):15345-15349 (1999)]. Carotenoids, especially lycopene, are particularly effective quenchers of singlet oxygen [ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS; Di Mascio,P; 274(2):532-538 (1989)].
Collagen & elastin also cross-link in skin, resulting in a loss of elasticity. The protein keratin in fingernails is also a component of the outer layer of skin (epidermis), which provides "water-proofing". The epidermis thins with age, leading to wrinkles. Decreased secretion by sweat glands increases vulnerability to heat stroke. When the melanocytes (cells that produce the skin&hair-coloring substance melanin) associated with hair follicles cease functioning, hair turns white. Partial reduction of melanocyte function results in hair that appears "gray". Yet 90% of Caucasians show increased melanin in the form of brownish spots on the back of their hands ("liver spots").
Although heat waves tend to lead to increased mortality among the elderly, those affected are generally persons with chronic disease conditions and unhealthy lifestyles. There is little alteration of thermoregulation with age among the normal elderly [JOURNAL OF APPLIED PHYSIOLOGY; Kenney;LW; 95(6):2598-2603 (2003)].
Loss of flexibility of the proteins collagen & elastin in the lung results in loss of elastic recoil. It becomes too difficult to fully exhale, which reduces air exchange, reducing the capacity to do work. Oxygen-to-tissue transfer rate is often halved by age 70.
Bone is typically 25% water, 30% soft tissue (cells & blood vessels) and 45% mineral deposits (mostly calcium). Most of the white ash remaining after cremation is calcium, lead, zinc and potassium from bone. Both men & women lose bone mass between the ages of 39 and 70 (osteoporosis), but post-menopausal women (who have reduce estrogen) lose bone mass at twice the rate as men. Decreased growth hormone causes bone loss in both sexes. The physical inactivity & malnutrition (especially for calcium and Vitamins D & C) of so many elderly also worsens bone loss. A reduction of one to three inches in height by age 80 is not unusual, with women shrinking twice as much as men. Young bones have been compared to green tree branches that can bend considerably before breaking — and upon breaking does so with splintering. By contrast, old bone is like a dry stick that snaps upon bending. 20% of hip fractures associated with osteoporosis are fatal in the US.
Joints in the bones of the inner ear calcify, contributing to a loss in the ability to hear higher tones. Loss of sweat glands in the ear causes earwax to become drier & crustier. Wax obstruction reduces the ability to hear low frequencies.
Aging reduces salivary secretion resulting in a drier mouth and decreased protection from bacterial infection of the mouth. Gastric juice volume is reduced 25% by age 60 and there is a 60% decline in pepsin activity. But this does not noticeably affect digestion except in the case of heavy meats. Absorption of Vitamin D (and, hence, calcium absorption), Vitamin B12 (affected by reduced "intrinsic factor") and folic acid all typically decline with age.
Atomic nuclei are surrounded by electron orbitals which contain a maximum of two electrons, each having opposite spin. Hydrogen has one outer orbital, but nitrogen, carbon and oxygen have 4 outer orbitals — with a capacity for 8 electrons (an "octet"). Atoms are most stable when they have filled orbitals. Free radicals are highly reactive molecules or atoms that have an unpaired electron in an outer orbital that is not contributing to molecular bonding ("free"). Atoms or small molecules that are free radicals tend to be the most unstable, because larger molecules can have the capacity to form resonance structures.
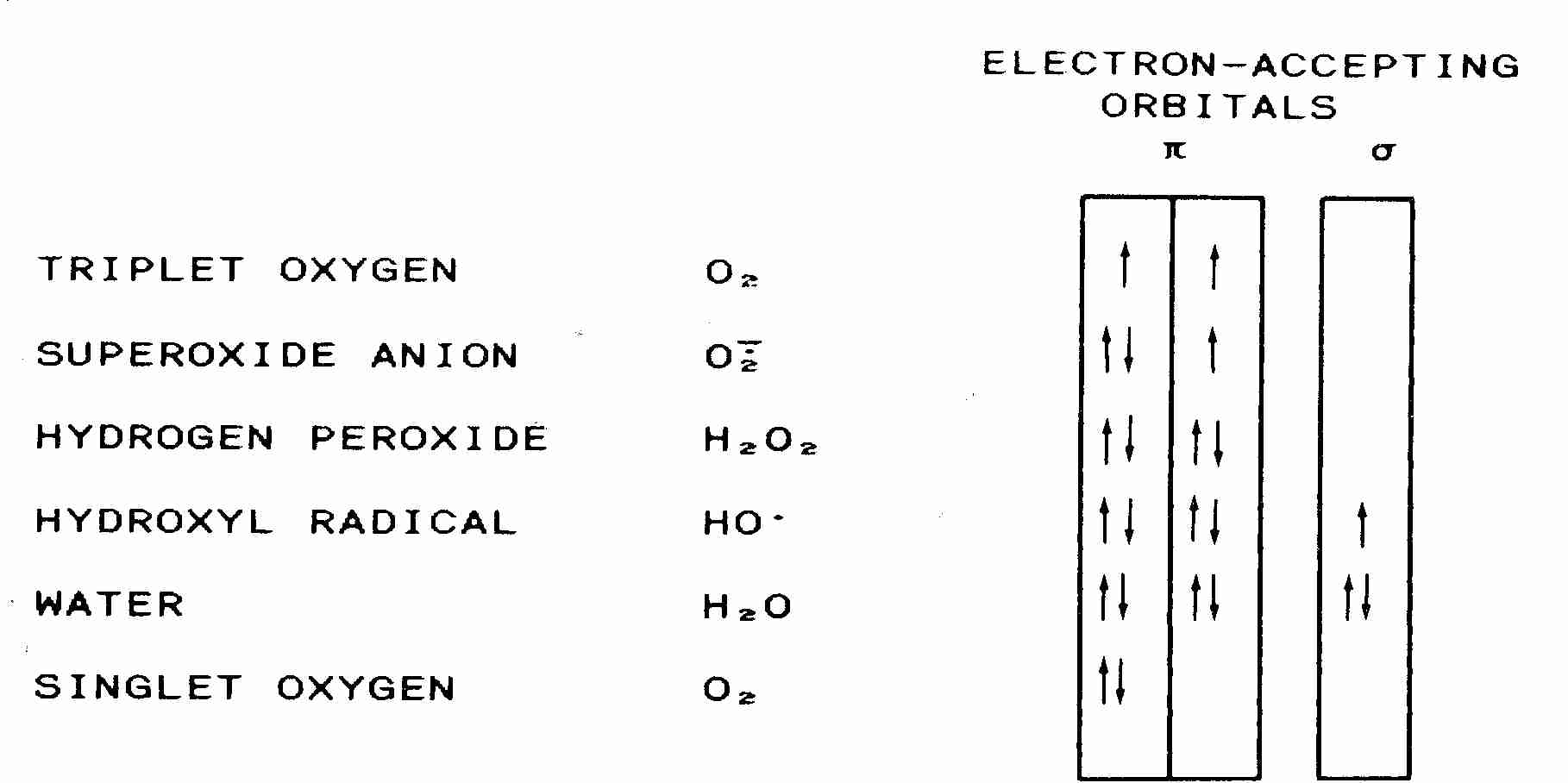
|
Normal molecular oxygen (3O2, so-called triplet oxygen) is a very unusual free-radical in that it has two unpaired electrons in outer orbitals (a double radical). Pi−bonds are bonds formed from overlapping p−orbitals. But for 3O2, two pi−bonds are formed from two p−orbitals, each containing one electron. The two electrons can have three possible arrangements: two "up"−spin (indicated by two up-arrows in the diagram), two "down"−spin or one spin "up" and one spin "down" — which makes 3O2 somewhat stable. But by the addition of energy (22.5 kcal/mole), both electrons are move into a single p−orbital, with the electrons having opposite spins — giving singlet oxygen (1O2).
Although singlet oxygen is not a free-radical, the electrons are in an excited state and can thus cause damaging reactions similar to those caused by oxygen free-radicals. On the other hand, if an electron is added to normal triplet oxygen, the new electron completes one orbital, leaving the other orbital with an unpaired electron — resulting in a superoxide anion (.O2−), which is a conventional, unitary free-radical. Singlet oxygen is attracted to double-bonds and can react destructively with DNA & proteins. Singlet oxygen is especially reactive with the amino acid histidine — resulting in enzyme denaturation. Singlet oxygen oxidizes the guanine base of DNA to produce 8−OHdG/8−oxoG [JOURNAL OF BIOLOGICAL CHEMISTRY; Ravanat,J; 275(51):40601-50604 (2000)]. Singlet oxygen from ultraviolet light is believed to be the major contributor to "photoaging" of the skin [JOURNAL OF BIOLOGICAL CHEMISTRY; Berneburg,M; 274(22):15345-15349 (1999)].
Lewis structures are structural chemical formulas depicting outer-shell electrons. I use abbreviated Lewis structures showing only relevant outer-shell electrons to explain free radicals — ie, I show a single orbital containing paired or unpaired electrons. Because an orbital containing one (unpaired) electron is not being complemented with an electron of opposite spin, the electron is said to be in an "unstable spin state" (another term for "free radical"). Thus, chemicals that react-with and stabilize free radicals are called spin-trapping substances.
Free radicals can damage nucleic acids, proteins or lipids. For biological systems, oxygen free radicals are the most important, in particular superoxide (.O2−), nitric oxide (.NO) and the hydroxyl radical (.OH). About 0.3% of superoxide exists in protonated form (HO2.), which is more reactive than superoxide itself. Because the protenated form of superoxide is uncharged, it can penetrate cell membranes more effectively than superoxide. Nitric oxide is a relatively unreactive free-radical which has a half-life of a few seconds, normally reacting quickly with oxygen (O2). But if nitric oxide encounters a superoxide (.O2−), it forms peroxynitrite (ONOO−) which can decompose to form a hydroxyl radical (.OH). Peroxynitrite, like the hydroxyl radical, can react directly with proteins and other macromolecules to produce carbonyls (aldehydes & ketones), cross-linking and lipid peroxidation. Only 1−4% of the DNA single-strand breaks caused by peroxynitrite are due to hydroxyl radical (indicating the minor effect decomposition has on total DNA damage by peroxynitrite) [ARCHIVES OF BIOCHEMISTRY AND BIOPHYSICS; Roussyn,I; 330(1):216-218 (1996)]. Although hydrogen peroxide (H2O2) and hypochlorite (OCl− — the active ingredient in bleach) are not themselves free radicals, these oxygen-containing molecules can facilitate free-radical formation. Moreover, HOCl is estimated to be hundreds of times more toxic than either hydrogen peroxide or superoxide [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Reiter,RJ; 917:376-386 (2000)].
All of these highly reactive oxygen-containing molecules (including singlet oxygen) are described as Reactive Oxygen Species (ROS). ROS attack bases in nucleic acids, amino acid side chains in proteins and double-bonds in unsaturated fatty acids — with the hydroxyl radical being the strongest attacker. ROS attack of macromolecules is often called oxidative stress. Reactive Nitrogen Species (RNS) also cause free radical damage. Peroxynitrite, which does most of its damage to endothelial cells, is nearly as destructive as the hydroxyl radical.
In a neutral water solution about one per 10−7 water molecules will dissociate into two ions, a reaction that can be represented as:
H:O:H => :OH− + H+
However, a water molecule subjected to ionizing radiation might dissociate into two free radicals: a hydroxyl radical & a hydrogen atom. The reaction can be represented as:
H:O:H => .OH + .H
A superoxide ion (.O2−) would result from the addition of an electron to a normal oxygen molecule (O2). A more complete Lewis structure of oxygen-containing free-radical molecules (with oxygen & hydroxide ion also illustrated for contrast) showing all outer shell electrons would be:
![[ oxygen free-radical molecules ]](radicals.gif)
It would be more accurate to draw resonance structures, but the above representations may be better for explanatory purposes.
The weed-killing herbicide paraquat generates superoxide. Superoxide (.O2−) ions are generated in large numbers in the mitochondria. Two superoxide ions are enzymatically converted to hydrogen peroxide (H2O2) by the enzyme superoxide dismutase:
.O2− + .O2− + 2H+ => H2O2 + O2
The hydroxyl radical (.OH) is typically formed by oxidation of a reduced heavy metal ion (Fe++ or Cu+, usually) by the hydrogen peroxide:
Fe++ + H2O2 => Fe+++ + .OH + :OH−
The last reaction, known as the Fenton Reaction, may be the most dangerous because it can occur in the cell nucleus and lead to DNA damage. The oxidized iron (Fe+++) can then catalyze the Haber-Weiss Reaction between superoxide and hydrogen peroxide to produce more hydroxyl radicals:
.O2− + H2O2 => O2 + .OH + :OH−
At neutral pH the Haber-Weiss reaction occurs only to a negligible extent when no metal ion is available to act as a catalyst. In the human body ascorbic acid is normally beneficial rather than harmful because nearly all iron and copper ions are tightly bound to carrier proteins (transferrin for iron and cearuloplasmin for copper ions), but this is not the case in the Cerebral Spinal Fluid (CSF) or where there is cellular breakdown due to ischemic-reperfusion injury. Bacteria are rich in iron, which is why hydrogen peroxide from macrophages is such an effective bacterial killer.
Metal ions can also react with ascorbate (Vitamin C) to produce singlet oxygen (1O2) from normal triplet oxygen (3O2):
Cu++ + ascorbate + 3O2 => 1O2
Unlike iron, copper generates more singlet oxygen than hydroxyl radical upon its reaction with hydrogen peroxide.
Wherever it is produced, the hydroxyl radical is highly reactive and can cause covalent cross-linking or free-radical propagation in a wide variety of biological molecules. A cell's superoxide ions tend to be concentrated in the mitochondria because they are too reactive to travel very far in an unaltered state — and are much less frequently found in the nucleus than in the cytoplasm. Similarly, hydroxyl radicals (which have a billionth-of-a-second half-life) do not drift far from their site of formation. But hydrogen peroxide molecules are more stable and can drift across the nuclear membrane into the nucleus or near cell membranes where hydroxyl radicals can be generated when heavy metal ions are encountered. Hydrogen peroxide can damage proteins directly by the oxidation of −SH groups.
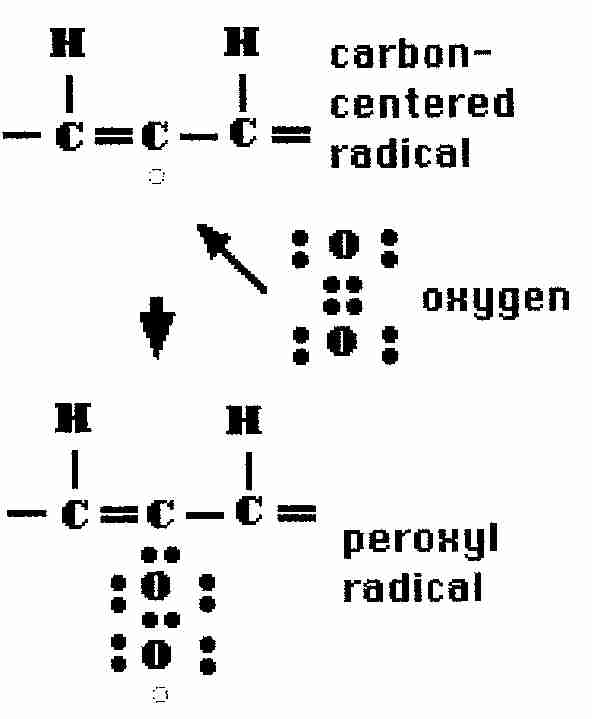
The hydroxyl radical can react with molecules (LH) in membranes to produce lipid molecule radicals (alkyl = .L)
|
.OH + LH => .L + H2O
These lipid radicals can then react directly with oxygen (autoxidation) in a self-propagating chain reaction forming lipid peroxides (lipid peroxyl radicals, lipid molecules containing paired-oxygen groups −−OO−−):
.L + O2
=> LOO.
LOO. + LH
=> LOOH + .L
The first reaction is about fifteen hundred times faster with singlet oxygen (1O2) than with normal triplet oxygen (3O2). Singlet oxygen is energetic enough, however, that it can react directly with the double bonds of unsaturated fatty acids, without requiring a free radical intermediate.
The lipid hydroperoxides (LOOH) can promote a Fenton reaction:
Fe++ + LOOH + H+ => Fe+++ + .OL + H2O
The lipid alkoxyl radical (alkoxy = alkoxyl = .OL) is more reactive and damaging than the lipid peroxide (peroxyl) radical (peroxy = peroxyl = LOO.). Thus, by a small sequence of steps one free-radical (.L) has become two radicals (.L and .OL) — conditions for an auto-amplifying chain reaction. Nonetheless, if two alkyl, alkoxyl or peroxyl radical molecules collide they will nullify each other, but at the cost of creating a cross-link (covalent bond) between the two lipids.
|
The reactivity of free radicals can be quantified by a table of half-life (time taken for half of the remaining radicals to react) values at 37ºC (body temperature). Short half-life corresponds to high reactivity. The one nanosecond half-life of the hydroxyl radical indicates that it is so reactive that it reacts with the first molecule it bumps into.
Outside of the mitochondria, superoxide and hydrogen peroxide can be generated on the endoplasmic reticulum through oxidation processes involving cytochrome P−450 and NADPH−cytochrome c reductase. Abnormal accumulation of normal metabolites such as lactate, pyruvate, acetoacetyl−CoA and glyceraldehyde−3−phosphate can abnormally increase levels of NADH oxidase & reduced flavoenzymes such as xanthine oxidase. In the absence of sufficient electron acceptor substrates these enzymes can directly transfer electrons to O2 or Fe+++ to form superoxide or Fe++. Ascorbate forms H2O2 on autoxidation (direct combination with oxygen). Both ascorbate & mercaptans (thioalcohols, ie, compounds having "−SH" groups, where sulfur is substituted for the oxygen of alcohol) are capable of reducing Fe+++ & Cu++ to Fe++ & Cu+, thereby promoting Fenton reactions.
Lipid peroxidation of polyunsaturated fatty acids exposed to oxygen leads
to rancidity in foods. In living animal cells peroxidized membranes lose
their permeability, becoming rigid, reactive and nonfunctional. Lipid peroxidation
can produce singlet oxygen, hydroperoxides and lipid epoxides. In addition, many
damaging aldehydes are formed during lipid peroxidation, particularly
MalonDiAldehyde (MDA, propanedial)
& 4−HydroxyNonEnal (4−HNE). MDA is a
major metabolite of
arachidonic acid (20:4)[fatty acid with 20−carbons &
4 double-bonds]. MDA assays (notably TBARS —
ThioBarbituric Acid-Reacting Substances)
have been widely used as a measure of cell membrane lipid peroxidation.
4−HNE is also a product of 20:4 fatty acid autoxidation. 4−HNE reacts
with cellular components more strongly than MDA. 4−HNE reacts readily
with histidine residues, sulfhydryl groups and primary amino groups of
proteins [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA);
Uchida,K; 89(10):4544-4548 (1992)]. The fact that 4−HNE is
the most toxic known aldehyde produced by lipid peroxidation (much more
toxic than MDA) and yet is practically non-reactive with TBA (about
95% of TDA reactivity is due to MDA) points to the deficiency of
TBARS as a lipid peroxidation assay [ALCOHOL & ALCOHOLISM
20(2):161-173 (1985)]. F2−isoprotanes, produced by
oxidation of arachidonic acid, are the best biomarkers of lipid
peroxidation [FASEB JOURNAL; Montuschi,P; 18(15):1791-1800 (2004)].
![[ LIPID PEROXIDATION FORMING MDA]](lipidmda.gif)
|
Unlike free-radicals, the aldehydes MDA, 4−HNE & other aldehydes are rather long-lived and can drift far from membranes, damaging a wide variety of proteins, lipids & nucleic acids [FREE RADICAL BIOLOGY AND MEDICINE 11:81-128 (1991)]. Such damaged molecules are called Advanced Lipid peroxidation End-products (ALE, which can be as resistant to degradation as AGEs [BRITISH JOURNAL OF PHARMACOLOGY; Negre-Salvayre,A; 153(1):6-20 (2008)]. 4−HNE inactivates glucose−6−phosphate dehydrogenase, an enzyme required for the formation of NADPH and for forming ribose residues for nucleic acid biosynthesis. Aldehyde-bridge formation leads to the protein-protein cross-linking associated with lipofuscin formation. Plasma levels of both MDA and 4−HNE rise significantly with age [FREE RADICAL RESEARCH; Gil,L; 40(5):495-505 (2006)].
Polyunsaturated fatty acids are more vulnerable to free radical oxidation than any other macromolecules in the body — and the sensitivity to free radical damage increases exponentially with the number of double bonds. Studies of the liver lipids of mammals & a bird (pigeon) show an inverse relationship between maximum lifespan and number of double bonds [JOURNAL OF GERONTOLOGY 55A(6):B286-B291 (2000)]. Nonetheless, brain phospholipid unsaturation does not vary much between mammals, probably indicating the importance of unsaturated fatty acids for neural function [COMPARATIVE BIOCHEMISTRY AND PHYSIOLOGY Part B 132:515-527 (2002)].
Animal cells contain three important enzymes to deal with the superoxide and hydrogen peroxide: SuperOxide Dismutase (SOD), glutathione peroxidase and CATalase (CAT). A dismutase is an enzyme that catalyzes the reaction of two identical molecules to produce molecules in different oxidative states. In the absense of SOD, two superoxide ions can spontaneously dismutate to produce hydrogen peroxide and singlet oxygen. SOD catalyzes a reaction between two superoxide ions to produce hydrogen peroxide and triplet oxygen.
Catalase catalyzes the formation of water & free oxygen from hydrogen peroxide. CAT is present in membrane-limited organelles known as peroxisomes. Peroxisomes contain enzymes that degrade amino acids & fatty acids — producing hydrogen peroxide as a byproduct.
![[FREE-RADICAL OXIDATION CHAIN]](oxidate.gif)
Glutathione is a tripeptide composed of the amino acids cysteine, glycine and glutamic acid. Glutathione is the major antioxidant in the non-lipid portion of cells (most of the cytoplasm). Glutathione exists in a reduced form (GSH) and an oxidized form (GSSG). Reduced glutathione hydrogen donation can neutralize a hydroxyl radical:
GSH + .OH —> .GS + H2O
and then oxidized glutathione radicals can neutralize each other:
.GH + .GH —> GSSG
Glutathione peroxidase neutralizes hydrogen peroxide by taking hydrogens from two GSH molecules — resulting in two H2O and one GSSG. The enzyme glutathione reductase then regenerates GSH from GSSG with NADPH as a source of hydrogen.
The elimination of hydrogen peroxide by glutathione can be written as the reaction:
2 GSH + H2O2 => GSSG + 2 H2O
Long-lived transgenic fruit flies in which the enzyme which synthesizes GSH was overexpressed showed a maximum lifespan extension of nearly 50% [JOURNAL OF BIOLOGICAL CHEMISTRY; Orr,WC; 280(45):37331-37338 (2005)]. Glutathione levels generally decline with age [JOURNAL OF ANTI-AGING MEDICINE; Lang,CA; 4(2):137-144 (2001)], although no reduction of serum glutathione was seen in elderly women deemed to be in excellent physical and mental health [JOURNAL OF LABORATORY AND CLINICAL MEDICINE; Lang,CA; 140(6):413-417 (2002)]. Free radicals act on lipids to produce peroxides (−O−O− bonds) resulting in mutagenic epoxides and insoluble & non-digestible age pigments such as lipofuscin. Glutathione peroxidase/glutathione destroys fat peroxides in the same way it eliminates hydrogen peroxide:
2 GSH + ROOH => GSSG + ROH + H2O
Superoxide dismutase(SOD) is the most abundant anti-oxidant enzyme in animals. The liver, in particular, is very high in SOD. Cellular concentration of SOD relative to metabolic activity is a very good lifespan predictor of animal species. Most mammals experience a lifetime energy expenditure of 200,000 calories per gram, but humans have an amazing 800,000 calories per gram. Humans have the highest levels of SOD — relative to metabolic rate — of all species studied. Oxidative damage to DNA is ten times greater in rats than in humans. Maximum lifespan correlates with lower rate of free-radical production and higher rate of DNA repair [JOURNAL OF COMPARATIVE PHYSIOLOGY B 168(3):149-158 (1998)].
The SOD molecule in the cytoplasm (SOD1) and outside of cells (SOD3) contains copper & zinc atoms (Cu/Zn−SOD), whereas the SOD in mitochondria (SOD2) contains manganese (Mn−SOD).
Superoxide dismutase without glutathione peroxidase or catalase (CAT) to remove hydrogen peroxide is of little value. Insects lack glutathione peroxidase, but experiments have been performed on fruit flies made transgenic by having extra genes for SOD, CAT or both. The flies that were given extra genes for SOD or CAT (but not both) had no more than a 10% increase in mean lifespan, with no increase in maximum lifespan. But flies that had extra genes for both SOD and CAT showed maximum lifespan increase by as much as a third, while showing less protein oxidative damage and better physical performance [SCIENCE 263:1128-1130 (1994)]. But criticisms that the above experiments had been performed on short-lived strains of flies led to later experiments on long-lived strains of flies which showed no lifespan extension for overexpression of Cu/Zn−SOD, Mn−SOD, catalase and thioredoxin [JOURNAL OF BIOLOGICAL CHEMISTRY; Orr,WC; 278(29):26418-26422 (2003)].
Nonetheless, an experiment using SOD/CAT mimetics in nematode worms increased mean lifespan 44% [SCIENCE 289:1567-1569 (2000)]. Selective inbreeding of bread-mold fungus resulted in strains with lifespans more than 6 times longer than wild-type — a change that was shown to be due to increased expression of antioxidant enzymes [FREE RADICAL BIOLOGY & MEDICINE 8:355-361 (1990)]. Females express both more Mn−SOD and more glutathione peroxidase than males, and this has been suggested to be the reason females live longer than males in mammalian species [FEBS LETTERS; Vina,J; 579(12):2541-2545 (2005)]. The maximum lifespan of transgenic mice has been extended about 20% by overexpression of human catalase targeted to mitochondria [SCIENCE; Schriner,SE; 308:1909-1911 (2005)]. Although naked mole rats exhibit high levels of oxidative damage, these levels remain unchanged for over two decades [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Perez,VI; 106(9):3059-3064 (2009)].
Radiation produces the hydroxyl radical, but most of the oxygen free radicals are byproducts of cell metabolism — particularly in the mitochondria, the lysosomes and the peroxisomes. One of the reasons these organelles are surrounded by membranes may be to protect the cell from the free-radicals they generate. DNA may be sequestered in the nucleus, in part, as additional protection against free radicals. Nonetheless, free radicals contribute to DNA damage and mutation.
In addition to enzymes, the animal cell uses many other chemicals to protect against oxygen free-radicals. Vitamin E is the main free-radical trap in the (lipid) membranes. Vitamin C acts as an anti-oxidant in the non-lipid ("watery") portions of cells, between cells and in the bloodstream. Melatonin, a hormone produced by the pineal gland in decreasing quantities with aging, efficiently crosses membranes (including the nucleus) and is effective against hydroxyl radicals.
Uric acid (which is mostly formed from purine degradation) protects Vitamin C from oxidation by divalent ions and can act as an anti-oxidant. Uric acid also protects against free-radical catalysis by binding iron. Humans have higher levels of uric acid than monkeys and other mammals because humans lack the enzyme uricase. But birds typically have twice the plasma uric acid concentration as humans. Birds often live several times as long as comparably sized mammals despite over twice the metabolic rate, 2−6 times the plasma glucose and a 3ºC higher body temperature.
Mammals fed anti-oxidants show up to a 30% increase in average lifespan, but no increase in maximum lifespan. Anti-oxidants are most valuable for animals that are cancer-prone, or subjected to radiation or chemical toxins. There are evidently homeostatic mechanisms in cells that govern the amount of allowable anti-oxidant activity. For example, increased levels of Vitamin E in the diet correlates with reduced levels of glutathione peroxidase activity, and vice versa. Vitamin E was shown to increase catalase in banana fruit-flies — with increasing doses of Vitamin E extending fruit-fly lifespan up to a dose of 5 micrograms/mL, above which increasing doses decreased lifespan [GERONTOLOGY 42:312-321 (1996)].
(For more on anti-oxidants and anti-oxidant enzymes, see my essay General Anti-Oxidant Actions.)
|
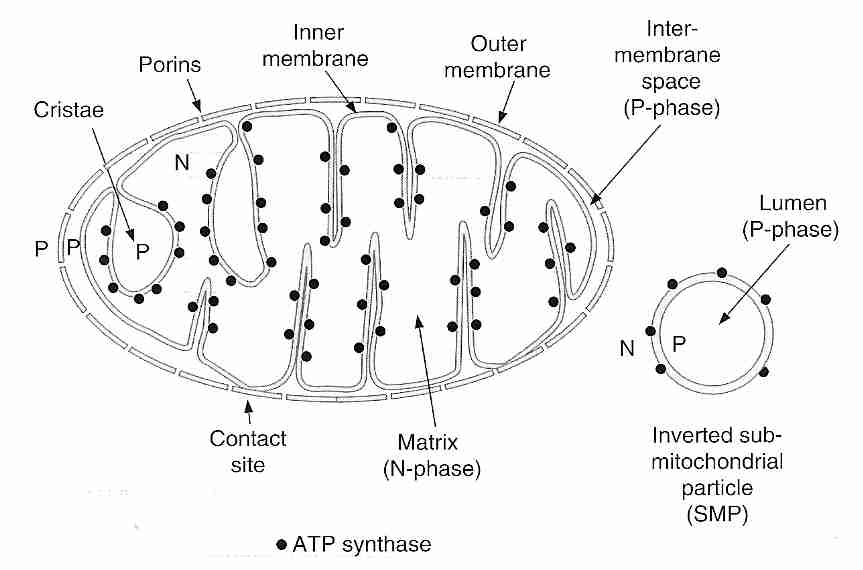
The mitochondria are capsule-shaped cellular organelles that generate energy (ATP molecules) from aerobic (oxygen-utilizing) metabolism utilizing respiratory chain and ATP synthase enzymes. Most animal cells contain between a few hundred and a few thousand mitochondria. The most mitochondria are found in the cells that are most metabolically active: neurons and muscle cells, where mitochondria make up about 40% of cell volume. About 10% of the body weight of a human adult is mitochondria.
A mitochondrion has two membranes. The outer membrane contains small pores (porins, also known as Voltage-Dependent Anion Channels,VDACs) that are freely permeable to ions and other molecules smaller than 10 kiloDaltons in size. The inner membrane is highly impermeable, even to protons (H+ ions). The proton gradient across the inner membrane is used by ATP synthetase enzyme to generate ATP molecules. The region between the outer membrane and the inner membrane is more positively charged (P−phase) because of the higher proton concentration, whereas the inside of the inner membrane is more negatively charged (N−phase, the matrix). It is in the matrix that the Krebs citric acid cycle occurs. There can be tens of thousands of respiratory chain and associated ATP synthase molecules embedded in the inner membrane of a mitochondrion, especially in metabolically active cells that have their inner membranes most highly folded into cristae that increase surface area.
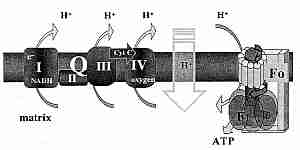
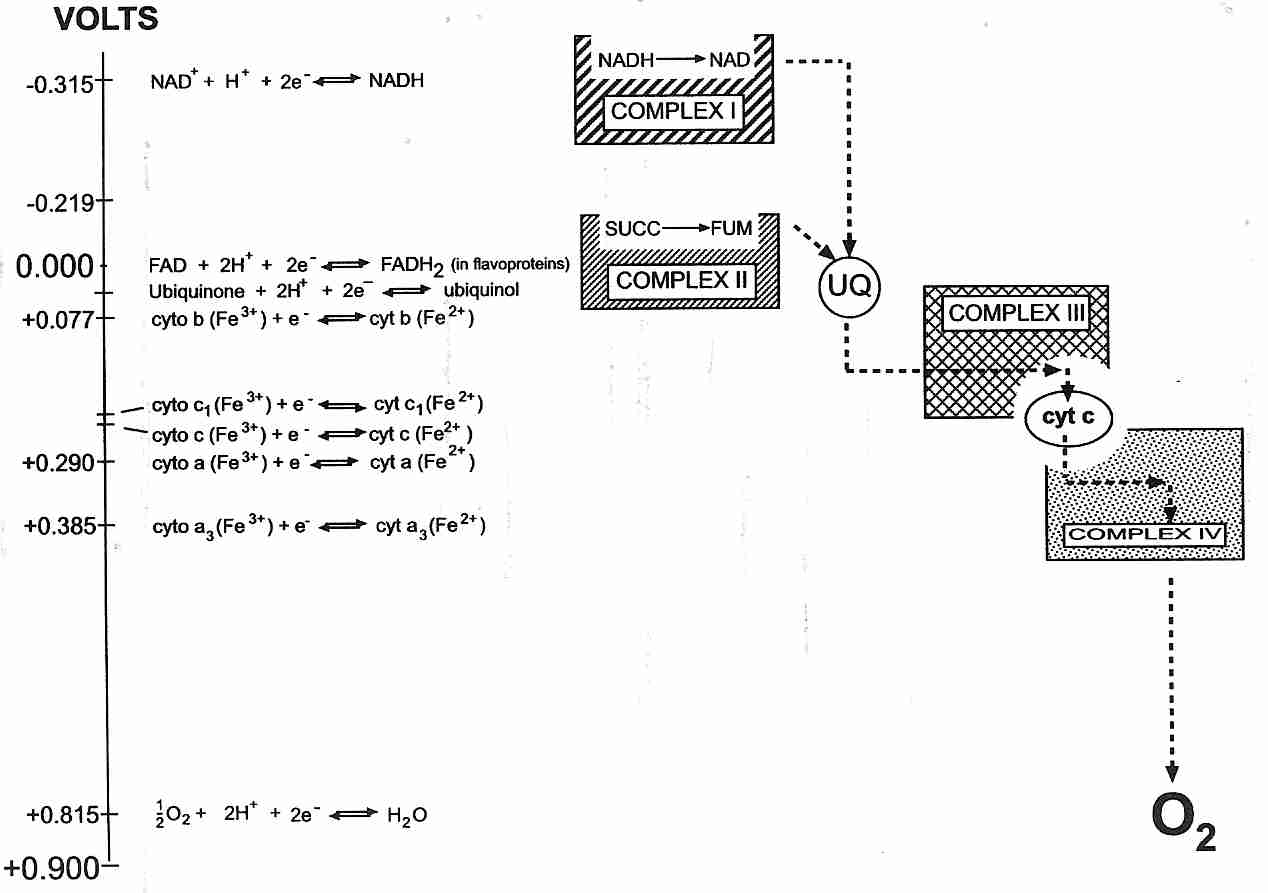
The inner membrane contains a number of active molecule carriers, including a phosphate (Pi = H2PO4-) carrier and the Adenine Nucleotide Transporter (ANT). The ANT imports ADP molecules into the matrix for ATP synthesis in exchange for ATP molecules which are exported for energy use throughout the cell (like portable batteries). The respiratory chain ("electron transport chain") attached to the inner wall of the inner membrane is composed of 4 protein complexes. These protein complexes are identified as Complex I, II, III and IV. Complex II consists of only four peptides, two of which comprise the Krebs citric acid cycle protein succinate dehydrogenase, and two of which anchor the complex to the inner mitochondrial membrane.
Complex I and Complex II independently supply electrons to Complex III, which supplies electrons to Complex IV. Soluble carriers are used to transport electrons to and from Complex III. The soluble carrier transporting electrons from Complex I & II to Complex III is Coenzyme Q (CoQ). The soluble carrier that transports electrons from Complex III to Complex IV is cytochrome−c. For this reason Complex III is also known as cytochrome−c reductase and Complex IV is also known as cytochrome−c oxidase. Complex IV combines its electrons (which are actually hydrogen atoms) with oxygen to form water. The energy released by the oxidations in the respiratory chain are used to pump protons outside the inner mitochondrial membrane.
|
The inner mitochondrial membrane is fairly impermeable to H+ ions ("protons") and thus is able to function much like a hydroelectric dam. Respiratory enzymes (Complex I, III & IV) pump protons out of the inner mitochondrial matrix, building proton pressure outside the "dam" (the membrane). The proton pressure ("proton-motive force") across the inner membrane is composed of two components: a pH difference and an electrical potential (membrane potential), which is the most important component. The pH difference is small, amounting to only about 0.5 pH units. The membrane potential of the mitochondrial membrane is about twice as great as that of a large nerve fiber, amounting to over 200 milliVolts. Complex V (F0F1−ATP synthase) is the "hydroelectric turbine" that utilizes the energy of the proton flow into the matrix through the "turbine" to synthesize ATP. The ATP synthase (Complex V) "rotary motor" is the smallest known natural nanomachine. It uses proton-motive force to drive the endothermic reaction:
ADP + Pi => ATP
The combined result of respiratory (oxidative) steps and the ATP-creation (phosphorylation of ADP) step is called oxidative phosphorylation. Normally respiration (oxygen consumption) and phosphorylation (ATP production) are tightly coupled, ie, the amount of ATP produced corresponds to the amount of oxygen consumed — referred to as state 3 respiration. In the absence of ADP (eg, in a resting state), however, any respiration that occurs will be due to "proton leak" through the inner mitochondrial membrane rather than due to ATP production — referred to as state 4 respiration. (State 1, state 2 and state 5 are experimental conditions of more historical interest than metabolic interest.)
In state 4 respiration protons flowing directly through the inner membrane rather than through the "ATP turbine" (Complex V) produce heat energy rather than ATP energy. Uncoupling proteins are weak acids that dissolve inner membrane lipids thereby increasing the uncoupling of oxidation from phosphorylation. Uncoupling respiration from phosphorylation to produce heat is useful for small rodents, naked newborn babies, and hibernating & cold-acclimated animals, all of which contain "brown fat". Uncoupling is also useful for fever production. UCP1 is the UnCoupling Protein found in "brown fat", fat which has been made brown by high concentrations of mitochondria. UCP2 has broad tissue distribution and seems to function in stress response, but its expression is less than 1% of UCP1. UCP3 is found in muscle and is regulated by thyroid hormone (T3). UCP2 & UCP3 may cause uncoupling for the purpose of reducing mitochondrial superoxide production [FREE RADICAL BIOLOGY & MEDICINE; Echtay,KS; 43(10):1351-1371 (2007)].
The function of UCP1 is to generate heat ("thermogenesis"). Claims have been made that UCP3 generates little heat, but functions to reduce free radical damage by lowering protein gratient during periods of high metabolic activity. Mice with higher UCP3 have shown higher metabolic intensity (17% greater resting oxygen consumption) and 36% longer lifespan [AGING CELL; Speakman,JR; 3(3):87-95 (2004)]. Proton leak has not been shown to be a factor in CRAN (Caloric Restriction with Adequate Nutrition) [AMERICIAN JOURNAL OF PHYSIOLOGY; Ramsey,JJ; 286(1):E31-E40 (2004)]. The fact that dieting-resistant obese subjects have been shown to have smaller amounts of UCP3 [DIABETES; Harper,M; 51(8):2459-2466 (2002)] would seem to indicate that thermogenesis from UCP3 is not negligible.
Compared to the heart & brain, mitochondria in the liver are more tightly coupled and use oxygen more efficiently for ATP production. The heart & brain mitochondria use more oxygen than liver mitochondria, but can produce ATP faster. Brain mitochondria are more geared toward maintaining cell integrity, in contrast to heart mitochondria which are more geared toward preserving cellular energy state [AMERICAN JOURNAL OF PHYSIOLOGY; Cairns,CB; 274(5):R1376-R1383 (1998)].
Increasing insulin levels associated with aging and type−2 diabetes stimulates nitric oxide synthetase resulting in peroxynitrite [THE INTERNATIONAL JOURNAL OF BIOCHEMISTRY & CELL BIOLOGY 34:1340-1354 (2002)]. Lipid peroxidation of the inner mitochondrial membrane by peroxynitrite can increase proton leak independent of uncoupling protein. Peroxynitrite can also degrade function of respiratory enzymes [JOURNAL OF NEUROCHEMISTRY 70:2195-2202 (1998)] and inactivate mitochondrial superoxide dismutase (Mn−SOD) enzyme [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA) 93(21):11853-11858 (1996)].
Mitochondria are the only
cellular organelles with their own DNA. (There is no other cellular DNA
outside the nucleus apart from the DNA of mitochondria.) Mitochondrial DNA
(mtDNA) in humans are circular strands of 16,569 nucleic acids that
code for 37 genes — 22 transfer RNAs, 2 ribosomal RNAs and 13 transmembrane
proteins. There are nearly 1,500 other gene products in mitochondria, which are
coded-for by nuclear DNA (nDNA). In contrast to nDNA, the mtDNA is
derived almost entirely from the mother. Each cell contains many mitochondria, but the total
mtDNA in a cell represents less than 1% of the amount of DNA found in the nucleus.
|
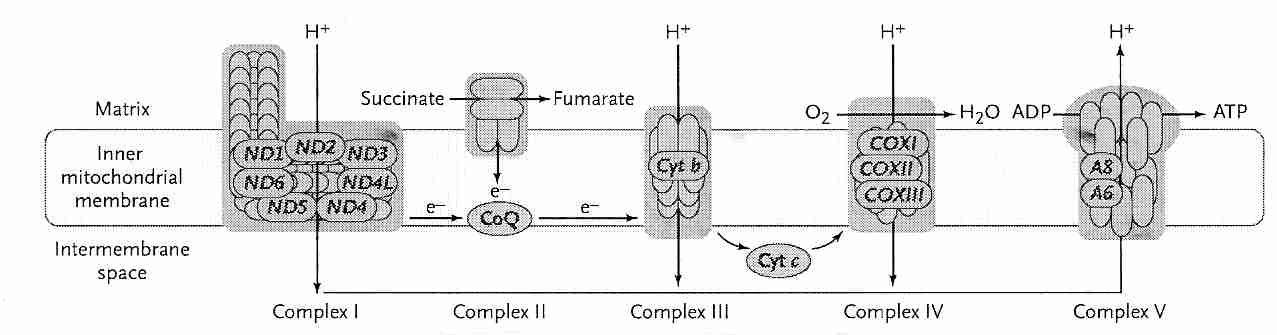
Each mitochondrion contains 2-to-12 identical copies of mitochondrial DNA (2-to-12 circular strands). Each mtDNA strand codes for 13 proteins, all of which are transmembrane subunits of Complex I, III, IV or V. Of the 13 mtDNA proteins, 7 are in Complex I, 1 is in Complex III, 3 are in Complex IV and 2 are in Complex V. A distinctive feature of the 13 proteins coded-for by the mtDNA is that they are hydrophobic (not easily dissolved in water), suggesting that it might be difficult to synthesize & transport them in the watery cytoplasm. For this reason it has seemed improbable that the mtDNA for these proteins could be moved to the nucleus where they would be better protected & repaired. But one of the Complex V (ATPase) mtDNA-coded proteins has been successfully synthesized in the nucleus and utilized in the mitochondria for a mammalian cell [REJUVENATION RESEARCH; Zullo,SJ; 8(1):18-28 (2005)] giving hope to the idea that all 13 mtDNA proteins might eventually be moved to the nucleus. An alternate hypothesis, however, claims that the mtDNA genes are of value in providing rapid local synthesis of proteins required for oxidative phosphorylation. Oxidative stress due to insufficient oxidative phosphorylation capability could signal mitochondrial transcription factors to induce production of mtDNA-coded proteins that are then implanted into the inner membrane where they attract the nDNA-coded proteins required for complete assembly of the complexes [PHILOSOPHICAL TRANSACTIONS OF THE ROYAL SOCIETY; Allen,JF; 358(1429):19-38 (2003)].
Complex I, which has 7 mtDNA-coded proteins (more than a quarter of all the proteins in the Complex), ages most rapidly. Substantia nigra neurons have increased susceptibility to Complex I defects — which may be responsible for Parkinson's Disease [NEUROBIOLOGY OF AGING; Smigrodzki,R; 25:1273-1281 (2004)]. By contrast, Complex II (which has no mtDNA-coded proteins) and Complex III (which has only one) are relatively unaffected by aging. Cytochrome−c oxidase (between Complex III and Complex IV) activity declines with age, resulting in increased production of superoxide and hydrogen peroxide. Diseases due to mutated mtDNA have the greatest effect on cells producing the most energy — cells of brain and muscle — hence mitochondrial diseases are often encephalomyopathies . A very common syndrome of mitochondrial disease is Mitochondria Encephalomyopathy, Lactic Acidosis & Stroke (MELAS). Homoplasmy describes the original condition of all of a person's mtDNA being the same, but as mtDNA mutations occur and the mutated mtDNA replicates, cells, tissues and even mitochondria can have a mixture of mtDNA types, a condition known as heteroplasmy.
An estimated 1−2% of oxygen used by mitochondria will normally "leak" from the respiratory chain to form superoxide [JOURNAL OF NEUROCHEMISTRY 59:1609-1623 (1992) & JOURNAL OF INTERNAL MEDICINE 238:405-421 (1995)]. The pro-inflammatory cytokine Tumor Necrosis Factor−alpha (TNF−α, associated with the metabolic syndrome) induces increased free radical production from the respiratory chain [AMERICAN JOURNAL OF RESPIRATORY CELL AND MOLECULAR BIOLOGY; Corda,S; 24(6):762-768 (2001)]. Aging is associated with decreased oxidative phosphorylation coupling efficiency and increased superoxide production. Free radicals can damage the mitochondrial inner membrane, creating a positive feedback-loop for increased free-radical creation. The "viscious cycle" theory that free radical damage to mitochondrial DNA leads to mitochondria that produce more superoxide has been questioned. The most damaged mitochondria are consumed by lysosomes whereas the more defective mitochondria (which produce less ATP as well as less superoxide) remain to reproduce themselves [REJUVENATION RESEARCH; de Grey,A; 8(1):13-17 (2005)]. But the efficiency of lysosomes to consume malfunctioning mitochondria declines with age, resulting in more mitochondria producing higher levels of superoxide. Mitochondria of older organisms are fewer in number, larger in size and less efficient (produce less energy & more superoxide).
Coenzyme Q (CoQ, in humans CoQ10) is also known as ubiquinone, so-called because it is "ubiquitous" (universally-found) in almost all cellular organisms, with the exception of gram-positive bacteria and some fungi. CoQ is an essential component of the mitochondrial respiratory chain. From Complex I or Complex II dehydrogenase CoQ is reduced to CoQH2 and subsequently oxidized in two steps — first to .CoQ− and then to CoQ. But .CoQ− is unstable and can easily errantly transfer an electron to an O2 molecule resulting in superoxide ion (.O2−).
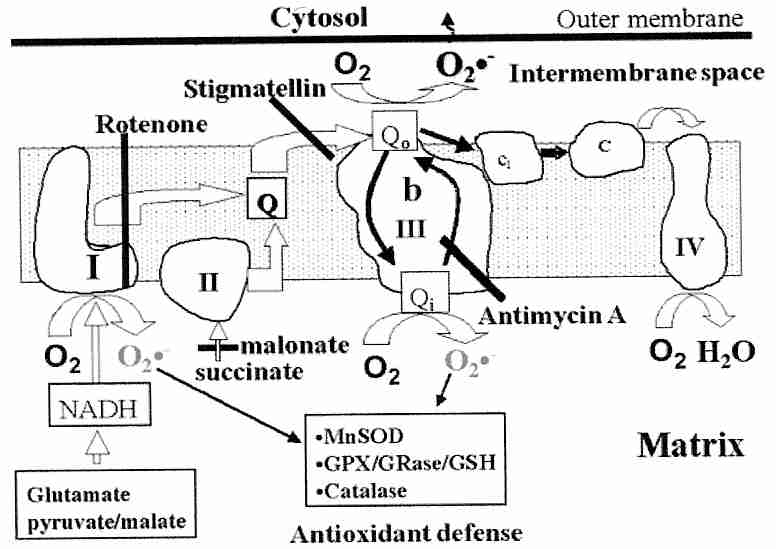
|
Complex I has been believed to generate .O2−
in one of the iron-sulfur clusters, which would go to the mitochondrial matrix
where it could be neutralized by Mn−SOD. Experiments on isolated mitochondria identified the
site of superoxide generation to be at the flavine mononucleotide moiety of
Complex I [JOURNAL OF BIOLOGICAL CHEMISTRY; Kudin,AP; 279(6):4127-4135 (2004)],
but claims have been made that experiments on isolated mitochondria are
misleading [ACTA BIOCHEMICA POLONICA; Nohl,H; 51(1):223-229 (2004)]. An experiment on
isolated synaptosomes indicated that Complex I inhibition
increases H2O2
production [THE JOURNAL OF NEUROSCIENCE; Tretter,L; 24(36):7771-7778 (2004)]. Most of the
.O2− generated from Complex III comes from
.CoQ−, with about half going to the matrix to be neutralized
and half floating toward the
cytoplasm [JOURNAL OF BIOLOGICAL CHEMISTRY; Muller,FL; 279(47):49064-49073 (2004)]. Thus,
.O2− from Complex I & III can cause
lipid peroxidation of the inner mitochondrial membrane and mtDNA damage, whereas
.O2− from Complex III can damage the whole
cell, including nDNA. Membrane potentials below 140 mV (potential
resulting from the proton gradients across the inner mitochondrial membrane) are not
associated with .O2−, but
above 140 mV .O2− generation
increases exponentially with potential. Uncoupling proteins can be a device
for reducing proton pressure (membrane potential), thereby reducing superoxide production.
|
Higher voltage drops between energy states in the Complexes also result in greater capacity for superoxide generation. This may account for the high superoxide production associated with Complex I, which has a high voltage drop in transferring its electrons to Complex III.
Oxidative damage to particular mitochondrial proteins in the flight muscles of houseflies has been identified as a biomarker of aging for those insects. Specifically, adenine nucleotide transferase enzyme in mitochondrial membranes [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Yan,L; 95(22):12896-12901 (1998)] and the citric acid cycle enzyme aconitase [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Yan,L; 94(21):11168-11172 (1997)] are particularly vulnerable to oxidative damage and are used to identify the "physiological age" of houseflies. Aconitase also shows the most significant age-related decline of any citric acid cycle enzyme in mice [MECHANISMS OF AGING AND DEVELOPMENT; Yarian,CS; 127(1):79-84 (2006)]. Aconitase is readily oxidized by superoxide, a process that generates hydroxyl radical [JOURNAL OF BIOLOGICAL CHEMISTRY; Vasquez-Vivar,J; 275(19):14064-14069 (2000)].
CoQ forms an important part of the antioxidant defense against these superoxide radicals [BIOCHEMISTRY AND CELL BIOLOGY 70:390-403 (1992)]. The Mn−SOD (SuperOxide Dismutase) of mitochondria can be induced to higher concentrations by oxidative stress (in contrast to the cytoplasmic Cu/Zn−SOD which is constitutive rather than induced). Heart mitochondria also contains catalase (which is confined to peroxisomes in most other tissues) [BIOSCIENCE REPORTS 17(1):3-8 (1997)].
Associated with aging is a decline in the amount of CoQ in organs. A person 80 years old will typically have about half as much CoQ10 in the heart, lungs and spleen as a 20-year-old [LIPIDS 24(7):579-584 (1989)]. Declines in functional mitochondria & CoQ10 with age is most damaging to those organs that have the highest energy demands per gram of tissue, namely: the heart, kidney, brain, liver and skeletal muscle, in that order [JOURNAL OF INTERNAL MEDICINE 238:405-421 (1995)]. Neurons are the largest cells in the body and have the highest metabolic demands, with 70% of ATP produced required to maintain the sodium-potassium pump. Clinically, damage to brain and muscle tissue are the first symptoms of mitochondrial disease. Mitochondria in the brain tissue of Alzheimer's Disease patients is particularly damaged. Therapy has included the B−vitamins that act as coenzymes in the respiratory chain (thiamine, riboflavin, niacinamide) and CoQ10 [ACTA NEUROLOGICA SCANDINAVIA 92:273-280 (1995)].
mtDNA deletion mutations accumulate in post-mitotic cells with age [BIOCHIMICA ET BIOPHYSICA ACTA 410:183-193 (1999)]. The "mitochondrial theory of aging" postulates that damage to mtDNA and organelles by free radicals leads to loss of mitochondrial function and loss of cellular energy (with loss of cellular function). Mutations in mtDNA occur at 10-20 times the rate seen in nuclear DNA. A significant portion of "photoaging" of the skin may be due to mtDNA deletions from singlet oxygen induced by ultraviolet light [JOURNAL OF BIOLOGICAL CHEMISTRY; Berneburg,M; 274(22):15345-15349 (1999)]. Transgenic mice having high levels of mtDNA point mutations and deletions are models of accelerated aging [CELL METABOLISM; Edgar,D; 10(2):131-138 (2009) and AGING; Edgar,D; 1(12):1028-1032 (2009)]. Unlike nuclear DNA, mtDNA has no protective histone proteins. And DNA repair is less efficient in mitochondria than in the nucleus. These factors account for the more rapid aging seen with Complex I & III as compared to Complex II & IV. Aging mitochondria become enlarged and, if they can be engulfed by lysosomes, are resistant to degredation and contribute to lipofuscin formation [EUROPEAN JOURNAL OF BIOCHEMISTRY; Brunk,UT; 269(8):1996-2002 (2002)].
A comparison of 7 non-primate mammals (mouse, hamster, rat, guinea-pig, rabbit, pig and cow) showed that the rate of mitochondrial superoxide and hydrogen peroxide production in heart & kidney were inversely correlated with maximum life span [FREE RADICAL BIOLOGY & MEDICINE 15:621-627 (1993)]. A similar study of 8 non-primate mammals showed a direct correlation between maximum lifespan and oxidative damage to mtDNA in heart & brain. There was a 4-fold difference in levels of oxidative damage and a 13-fold difference in longevity, supportive of the idea that mtDNA oxidative damage is but one of several causes of aging [THE FASEB JOURNAL; Barja,G; 14(2):312-318 (2000)].
A comparison of the heart mitochondria in rats (4-year lifespan) and pigeons (35-year lifespan) showed that pigeon mitochondria leak fewer free-radicals than rat mitochondria, despite the fact that both animals have similar metabolic rate and cardiac output. Pigeon heart mitochondria (Complexes I & III) showed a 4.6% free radical leak compared to a 16% free radical leak in rat heart mitochondria [MECHANISMS OF AGING AND DEVELOPMENT 98(2):95-111 (1997)]. Hummingbirds use thousands of calories in a day (more than most humans) and have relatively long lifespans (the broad-tailed hummingbird Selasphorus platycerus has a maximum lifespan in excess of 8 years). Birds have less unsaturation (oxidizability) in their mitochondrial membranes and have higher levels of small-molecule antioxidants, such as ascorbate & uric acid. Even for mammals there is a direct relationship between mitochondrial membrane saturation and maximum lifespan [JOURNAL OF LIPID RESEARCH; Pamplona,R; 39(10):1989-1994 (1998)].
Free-radicals from mitochondria result in damage to cellular protein, lipids and DNA throughout the cell. This damage has been implicated as a cause of aging. If the fatty acids entering the mitochondria for energy-yielding oxidation have been peroxidized in the blood, this places an additional burden on antioxidant defenses. The greatest damage occurs in the mitochondria themselves, including damage to the respiratory chain protein complexes (leading to higher levels of superoxide production), damage to the mitochondrial membrane (leading to membrane leakage of calcium ions and other substances) and damage to mitochondrial DNA (leading to further damage to mitochondrial protein complexes). An experiment in yeast that improved the accuracy of mitochondrial protein synthesis demonstrated a 27% longer mean life span [JOURNAL OF GERONTOLOGY 57A(1):B29-B36 (2002)].
|
Mitochondria play a key role in apoptosis ("cell suicide"). Release of cytochrome−c from mitochondria into the cytoplasm is the event which initiates apoptotic cell destruction by caspase enzymes. Release of cytochrome−c into the cytoplasm can occur either by a Ca2+−dependent mechanism or a Ca2+−independent mechanism. In the Ca2+−dependent case Ca2+ overload in the mitochondrion triggers opening of the Mitochondrial Permeability Transition Pore (MPTP), which penetrates both the outer and inner membranes making a channel between the mitochondrial matrix and the cytosol outside the mitochondrion. The MPTP is a complex consisting of three proteins, VDAC (porin) of the outer membrane, ANT (Adenine Nucleotide Translocator) of the inner membrane and cyclophilin−D. Cyclophilin−D protein binds to ANT to promote MPTP formation [BIOCHEMICAL JOURNAL; Li,Y; 383(Pt 1):101-109 (2004)], possibly by increasing the sensitivity of the MPTP components to the effects of Ca2+ [CIRCULATION RESEARCH; Weiss,JN; 93(4):292-301 (2003)]. The entry of large solutes and accompanying water into the matrix causes the mitochondrion to swell and burst, releasing cytochrome−c into the cytoplasm.
The Ca2+−independent case requires two
separate events for cytochrome−c release: (1) formation of large
pores in the outer mitochondrial membrane by Bax/Bak proteins and (2) release
of cytochrome−c from the inner mitochondrial
membrane [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA);
Ott,M; 99(3):1259-1263 (2002)]. The Ca2+−independent case
can lead to
apoptosis,
whereas the Ca2+−dependent case is invariably associated with
necrosis.
In apoptosis the MPTP opens only briefly (if it opens at all), whereas in necrosis
the MPTP remains open. Apoptosis requires ATP energy, but ATP energy is depleted
if the MPTP remains open [NATURE; Halestrap,A; 434:578-579 (2005)]. The threshold
amount of Ca2+ which causes MPTP opening in lymphocytes, brain and liver
of old mice is significantly lower than that of young mice [BIOCHEMICAL AND
BIOPHYSICAL RESEARCH COMMUNICATIONS; Mather,M; 273(2):603-608 (2000)].
CoEnzyme Q10
has been shown to reduce apoptosis
by direct inhibition of the MPTP [JOURNAL OF BIOLOGICAL CHEMISTRY; Papucci,L; 278(30):28220-28228 (2003)].
|
Cytochrome−c is normally held to the inner mitochondrial membrane by the lipid cardiolipin (diphosphatidylglycerol). Cardiolipin composes 10% of the inner mitochondrial membrane and is present at lower concentrations in the outer mitochondrial membrane (especially near contact sites between the two membranes). This distinctive lipid is found only in mitochondrial membranes. Mitochondrial membrane cardiolipin content declines with age, resulting in a decline in cytochrome−c activity. 40% lower cardiolipin content and 35% lower cytochrome−c activity has been demonstrated in old rats compared to young rats. Restoration of membrane cardiolipin content restored cytochrome−c activity [FEBS LETTERS; Paradies,G; 406(1-2):136-138 (1997)].
Oxidation of cardiolipin releases cytochrome−c from the inner mitochondrial membrane, but cytochrome−c will not be released into the cytoplasm to induce apoptosis without the formation of large pores in the outer mitochondrial membrane by Bax/Bak protein. Bax/Bak membrane permeabilization occurs preferentially at cardiolipin-rich contact sites between the outer and inner mitochondrial membrane [BMC CELL BIOLOGY; Lutter,M; 2:22-30 (2001)]. But Bax/Bak permeabilization of the outer membrane alone may be sufficient to induce apoptosis.
If only one or a few mitochondria release cytochrome−c apoptosis may not occur, but the damaged mitochondria would themselves be degraded. By this means a few aberrant mitochondria which are producing excessive free radicals can be eliminated.
(For more about mechanisms of apoptosis see
Cellular Senescence and Apoptosis in Aging)
Proteins are long chains of amino acids (amino acid polymers, or polypeptides). 20 different amino acids occur in animal proteins. Amino acids are all organic compounds with a protonated amino group [−NH3+] and an ionized carboxyl group [−COO−] attached to the same (alpha-position) carbon atom.
![[AMINO ACIDS]](amino.gif)
![[THE PEPTIDE BOND]](peptide.gif)
Proteins can be damaged both by free-radicals and by glycation. Glycation (also called the Maillard reaction, or non-enzymatic glycosylation) is a reaction by which reducing sugars become attached to proteins without the assistance of an enzyme. (For details on the properties of reducing sugars, see cryopreservation with sugars.) This attachment occurs at the free amine group of lysine or arginine, which is not involved in the peptide bond. The reaction between glucose and a lysine amino acid in a protein molecule can be represented as follows:
![[glycation/oxidation
reactions form AGEs ]](amadori.gif)
In the diagram, glycation is the formation of a double-bond between the glucose aldehyde-group and the lysine amino group with the elimination of a water molecule. The double-bond between the glucose carbon and the lysine nitrogen is an imine (also known as a Schiff base). The imine can quickly re-arrange atoms such that the 2-carbon (2nd carbon) of the glucose loses its two hydrogens — resulting in a carbonyl group (>C=O) and in hydrogen-saturation of the carbon & nitrogen which formerly constituted the imine. This re-arrangement structure is called an Amadori product (a ketoamine). Both glycation and Amadori product formation are completely reversible reactions. But the formation of Advanced Glycation End-products (AGEs) by oxidation of Amadori products is irreversible.
AGEs in tissues increase the rate of free radical production to 50-times the rate of free-radical production by unglycated proteins. Histochemical analysis of the hippocampus of human cadavers shows that chronological age can be estimated by hippocampal AGE levels [HISTOPATHOLOGY; Sato,Y; 38(3):217-220 (2001)]. AGEs attached to LDL-cholesterol accelerates oxidation and subsequent atherosclerosis. The irreversible cross-linked proteins of AGEs in vessel collagen also contributes to atherosclerosis, as well as to kidney failure — conditions worsened in diabetes [DIABETES 46(Suppl 2):S19-S25 (1997)]. Cataracts are composed of urea-insoluble proteins in the lens of the eye. AGEs aggravate protein cross-linking in the plaques & tangles of Alzheimer's Disease, thereby accelerating neuron death [BRAIN RESEARCH REVIEW 23:134-143 (1997)]. AGEs can be formed in the body from glycation & oxidation or can be ingested directly from browned foods (such as fried poultry skin) or tobacco smoke. Approximately one third of absorbed dietary AGEs are excreted in urine and rest is presumably incorporated into body tissues [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Koschinsky,T; 94(12):6474-6479 (1997)]. (For more on the effect of ingested AGEs, see INGESTION OF ADVANCED GLYCATION END-PRODUCTS (AGES).)
The higher glycation rate in diabetics is undoubtedly related to the fact that diabetes greatly resembles accelerated aging. Hemoglobin glycation is often used as a time-integrated (as opposed to instantaneous) measure of blood glucose levels in diabetics. AGEs are universal symptoms of aging — adversely affecting skin, lungs, muscles, blood vessels and organ-function in general. Increased insulin resistance and other symptoms of diabetes are commonly seen features of aging. Diabetes-like atherosclerosis and the resultant generalized reduction in blood flow has an adverse effect on most organ systems.
Although most proteins are short-lived (mouse liver proteins have a half-life of 3 days) some proteins, such as crystallins in the eye lens of mammals, can last a lifetime. Lens crystallines, collagen and basement membrane are the proteins most vulnerable to cross-linking and AGE formation because they are the most long-lived proteins, with a slow rate of turnover.
AGE cross-links can be broken by N−PhenacylThiazolium Bromide (PTB), but 3−phenacyl-4,5-dimethylthiazolum chloride (ALT−711, alagebrium) has proven to be even more effective than PTB in breaking cross-links [CIRCULATION RESEARCH; Candido,R; 92(7):785-792 (2003)]. Alagebrium has proven effective in reducing systolic blood pressure [AMERICAN JOURNAL OF HYPERTENSION; Bakris,GL; 17(12 Pt 2):23S-30S (2004)] and providing therapeutic benefit for patients with diastolic heart failure [JOURNAL OF CARDIAC FAILURE; Little,WC; 11(3):191-195 (2005)]. Carnosine also has anti-glycating effects [CURRENT MEDICINAL CHEMISTRY;Guiotto,A; 12(20):2293-2315 (2005)].
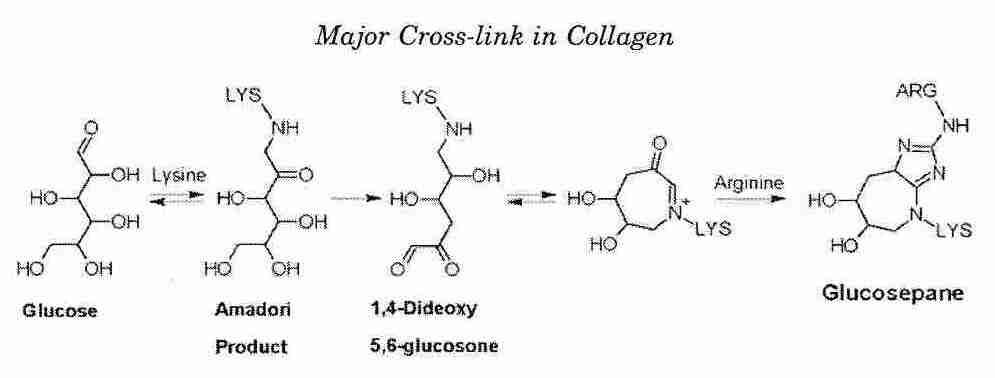
|
In the extracellular matrix of senescent skin, the major protein cross-link is the arginine-lysine glucose product glucosepane [REJUVENATION RESEARCH; Svantesson,J; 12(3):137-148 (2009)]. In non-diabetic 90-year-olds glucosepane accounts for about 50 times the cross-linking as all other forms of cross-linking, and is more than twice as prevalent in diabetics as in non-diabetics [JOURNAL OF BIOLOGICAL CHEMISTRY; Sell,DR; 280(13):12310-12315 (2005)]. In diabetics, prosclerotic growth factors like TGF−ß (Transforming Growth Factor beta) promote extracellular matrix synthesis.
Collagen accounts for about a third of total body protein in mammals. Collagen cross-linking in skin, muscle and organs throughout the body leads to the sinewy, inelastic tissue characteristic of aging. Cross-linking of proteins makes connective tissue lose elasticity, increases arteriosclerosis, reduces kidney function, slows wound healing, reduces the vital capacity of the lung and contributes to cataracts. Cross-linking also contributes to arteriosclerosis by making LDL-cholesterol unrecognizable to LDL-receptors, thereby increasing LDL in the blood.
Birds have blood glucose levels that are 2−10 times higher, metabolic rates that are more than double and body temperatures 2ºC−4ºC higher — than similarly-sized mammals. Higher temperatures & higher blood glucose would be expected to accelerate glycation & AGE formation in birds — yet their lifespans are considerably longer than those of comparably-sized mammals. Hummingbirds have the highest levels of glycated hemoglobin of any bird, but these levels are lower than those seen in non-diabetic humans, partially because of higher red blood cell turnover and partially because of better membrane control of glucose transport [COMPARATIVE BIOCHEMISTRY AND PHYSIOLOGY; Beuchat,CA; 120(Part A): 409-416 (1998)].
|
Birds have twice the blood concentration of antioxidant uric acid as humans and a much lower rate of free radical production. A study of hens showed less than one hundredth the quantity of Advanced Glycation End-products (AGES) as would be found in humans [JOURNALS OF GERONTOLOGY; Iqbal,M; 54A(4):B171-B176 (1999)]. The dipeptide carnosine (ß−alanyl-L-histidine) both inhibits glycation and has antioxidant metal chelating activity. Skeletal muscle concentrations of carnosine correlate with lifespan in mammals [BRAIN RESEARCH REVIEWS 23:134-143 (1997)]. Vitamin C also has anti-glycation properties as well as antioxidant action [DIABETES; Davie,SJ; 41(2):167-173 (1992)].
Glucose is not the most active sugar for glycation. Galactose is 5 times more reactive than glucose, fructose is 8 times more reactive, deoxyglucose is 25 times more reactive, ribose is 100 times more reactive and deoxyribose is 200 times more reactive. [Sucrose is composed of the two monosaccharides glucose & fructose, whereas lactose (milk-sugar) is composed of the two monosaccharides glucose & galactose.] Mice injected with galactose are models of accelerated aging [MECHANISMS OF AGING AND DEVELOPMENT; Song,X; 108(3):239-251 (1999)]. Some aldehydes produced by lipid peroxidation are more reactive than any of the sugars. Glucose assumes the cyclic conformation more readily than any other monosaccharide, making it the most resistant to both glycation and oxidation of any sugar. It is no evolutionary accident that the least reactive of sugars is the sugar organisms most use for energy.
Lipids as well as proteins are subject to glycation. Lipid glycation of LDL cholesterol increases the LDL oxidation associated with atherosclerosis [ATHEROSCLEROSIS, THROMBOSIS, AND VASCULAR BIOLOGY; Ravandi,A; 20(2):467-477 (2007)]. Vitamin B6 prevents lipid glycation more effectively than other common anti-glycation agents [JOURNAL OF LIPID RESEARCH; Higuchi,O; 47(5):964-974 (2006)].
(For more information on glycation, see the International Maillard Reaction Society.)
Not all of the damaging effects of sugar are due to glycation. Glucose & fructose are reduced to sorbitol by the enzyme aldol reductase. Sorbitol is a tissue toxin, contributing to retinopathy, neuropathy, cataracts and kidney disease in diabetes. And not all protein cross-linking is due to glycation. Aldehydes produced by lipid peroxidation, such as MalonDiAldehyde (MDA, propanedial), can cross-link proteins by forming covalent bonds with lysine amino acids.
| Cross-linking of protein with lipid peroxidation product MDA |
|---|
![[ Cross-linking of protein with lipid peroxidation product MDA ]](MalCross.gif)
|
Mitochondria produce nitric acid at a rate comparable to the rate of superoxide production [JOURNAL OF BIOLOGICAL CHEMISTRY; 273(18):11038-11043 (1998)]. Peroxynitrite formed by reaction of nitric oxide with superoxide can irreversibly form covalent bonds with tyrosine amino acids in proteins, thereby blocking phosphorylation. Phosphorylation & dephosphorylation of enzymes by kinases & phosphatases at tyrosine, serine & threonine protein residues play a central role in enzyme activation/deactivation & cell signalling — both of which would be disrupted by nitrotyrosine formation.
Another form of protein damage is racemization, although this kind of protein damage is less serious than glycation. Cells can only make proteins from L−isomer ("left-handed") amino acids. Only L−isomer proteins are functional. Some D−isomer ("right-handed") proteins are not only non-functional, but harmful. Thermal energy causes a small percentage of proteins to spontaneously change from the L−form to the D−form — and this form of molecular deterioration is known as racemization. (Racemization allows for determination of an animal's age from the ratio of D−form to L−form in the dentine of a tooth).
Asparagine and glutamine amino acids on proteins spontaneously deaminate, especially when a glycine is in the adjacent carboxyl position. The rate of deamination of asparagine is 400 times greater than for glutamine. When the enzyme which can repair this protein damage is missing from experimental (knockout) mice, the mice suffer brain damage and die young [JOURNAL OF BIOLOGICAL CHEMISTRY; 276(23):20695-20702 (2001)].
Carbonyl (>C=O, ie, aldehyde or ketone) content of protein is used as a rough measure of protein oxidation. Carbonyl formation is irreversible, so oxidized proteins must be removed by degradation. Carbonyl content of protein in an animal cell increases exponentially with age. At least 30−50% of protein is oxidized in old animals, which correlates well with an estimated 30−50% decrease in enzyme activity in old animals. A study on houseflies showed an association between protein carbonyls and life expectancy, possibly indicative of an effect on rate of aging [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Sohal,RS; 90(15):7255-7259 (1993)].
A study of several species of mammals & a bird (pigeon) indicated a linear relationship between oxidative damage to protein and maximum life span [EXPERIMENTAL GERONTOLOGY 31(3):365-372 (1996)]. Cysteine & methionine are by far more vulnerable to oxidation by reactive oxygen species than any other protein amino acids because of their sulfhydryl groups. Oxidation of cysteine sulfhydryls can result in disulfides that cause protein aggregation and lipofuscin. Naked mole rats (which live at least 9 times longer than mice) have much more oxidative damage to proteins than mice, but maintain that level of damage unchanged for two decades. Although mice have a 12% oxidative decline in cysteine with age, naked mole rats show no age-related change in cysteine for two decades [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Perez,VI; 106(9):3059-3064 (2009)].
Methionine is oxidized to methionine sulfoxide, but methionine sulfoxide reductases enzymatically regenerate methionine [BIOPHYSICA ET BIOCHEMICA ACTA; Lee,BC; 1790 (11): 1471-1477 (2009)]. Additionally, isomerases can reverse the aberrant disulfide bridges — the only known enzymatic repairs of protein oxidation [FREE RADICAL BIOLOGY & MEDICINE; Shringarpure,R; 32(11):1084-1089 (2002)]. Transgenic fruit flies that overexpress methionine sulfide reductase primarily in the nervous system have shown a median lifespan extension of about 70% [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); 99(5): 2748-2753 (2002)]. In one study, methionine sulfoxide reductase knockout-mice showed reduced lifespan and increased carbonyl content on protein [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); 98(23): 12920-12925 (2001)], but another study showed no reduction in lifespan [THE FASEB JOURNAL; Salmon,AB; 23(20):4601-3608 (2009)]. Reducing dietary methionine to a fifth the normal intake has increased the lifespan of rats by 30% [JOURNAL OF NUTRITION; Orentreich,N; 123(2):269-274 (1993)]. Body weight is just over half normal for the rats and there are increased blood as well as decreased tissue levels of glutathione [THE FASEB JOURNAL; Richie,JP; 8(15):1302-1307 (1994)].
Oxidized protein is more vulnerable to cross-linking by lipid peroxidation aldehydes such as MDA & 4−HNE [FEBS LETTERS 405:21-25 (1997)]. Hydroxyl radicals cause protein crosslinking by the formation of dityrosine bridges. Oxidized & cross-linked proteins are resistant to degradation and therefore contribute to the accumulation of damaged proteins in many degenerative diseases.
Cellular proteins are continually being degraded (hydrolyzed) within cells by proteolytic enzymes, both for regulation of cellular processes and for "quality control" of proteins (eliminating malformed or malfunctional ones). Transcription factors and cell-cycle proteins must be quickly eliminated after having served their purpose. The four major classes of cellular proteolytic enzymes are (1) caspases (2) calpains (3) cathepsins and (4) proteasomes.
Caspases are mainly active in apoptosis and are therefore in the category of regulatory proteases. Calpains are Ca2+−dependent, ATP-independent proteases that mainly degrade membrane & cytoskeletal proteins (as well as certain transcription factors). Cathepsins are the major class of proteolytic enzymes found in lysosomes, although there are others. (As well, lysosomes contain enzymes for degrading lipids, carbohydrates and nucleic acids.) Proteasomes are enzymatic, proteolytic "machines".
|
Normally, most damaged or misfolded protein — as well as obsolete regulatory protein — is eliminated by the barrel-shaped proteasomes, which are found both in the nucleus and in the cytoplasm. The core "barrel", called the 20S proteasome, is capped on one or both ends by 19S regulatory units, and the whole structure is called the 26S proteasome. Proteins are hydrolyzed inside the 20S proteasome core. The 19S regulatory units assist in recognition and delivery to the 20S core of proteins that have been marked for degradation by chains of ubiquitin (a 76-amino acid globular protein). Ubiquitin-activating enzymes use ATP to add ubiquitin to proteins requiring degradation. When the chains reach a threshold length of four ubiquitin subunits, the marked protein is hydrolyzed by the proteasome into reusable peptides and ubiquitin molecules. Oxidized proteins tend to be partially unfolded (denatured) and more hydrophobic, which may make them directly recognizable to the 20S cores, such that energy-consuming ubiquitination is not required for degradation.
Proteasome activity declines with age. Polyubiquinated chains of defective proteins bind to the 19S regulatory units blocking the passageway and preventing recognition of other ubiquinated proteins. Even when the 19S regulatory units are clear, accumulating large masses of cross-linked proteins cannot enter the proteasome, which has a 5−6 nanometer size opening.
Degradation of cellular organelles, proteins and other materials by lysosomes is called autophagy, subdivided into macroautophagy, microautophagy, and CMA. In macroautophagy there is a sequestration of complete portions of the cytosol (often including phagocytosed material or organelles) into a double membrane vesicle known as the autophagosome, which migrates to the lysosome and fuses with the lysosome membrane. In microautophagy the lysosome membrane itself engulfs portions of the cytoplasm. Mice with defective autophagy suffer neurodegeneration [NATURE; Hara,T; 441:885 (2006)] and DNA damage [GENES & DEVELOPMENT; Mathew,R; 21(11):1367-1381 (2007)]. Macroautophagy is mainly controlled by the kinase mammalian Target of Rapamycin (mTOR), a downstream component of the PI3K pathway, which is inhibited by rapamycin or absence of nutrition. Rapamycin extends both median and maximum lifespan when fed to mice [NATURE; Harrison,DE; 460:392-395 (2009)]
Proteins can also be brought into the lysosome for degradation by Chaperone-Mediated Autophagy (CMA) in which members of the hsp70 heat shock protein family (the chaperones) attach to a target protein and then bind to a lysosome receptor protein. CMA operates on proteins having the exposed KFERQ pentapeptide (K=Lysine, F=Phenylalanine, E=Glutamate, R=Arginine, D=Glutamine — IUPAC amino acid abbreviations) [MOLECULAR BIOLOGY OF THE CELL; Kiffin,R; 15(11):4829-2840 (2004)]. Upon arrival at the lysosome membrane, the chaparone/protein complex binds to the membrane protein LAMP−2a. The decline in CMA activity in aging is due to declining LAMP−2a in the lysosome membrane [NATURE MEDICINE; Zhang,C; 14(9):959-965 (2008)].
Proteins with short half-lives tend to be broken-down by the proteasome, whereas proteins with half-lives in excess of ten hours tend to be degraded by autophagy. Blockage of proteasomes by protein aggregates would result in cell dysfunction due to the inability to degrade short-lived regulatory proteins. Impaired degradation of p53 protein can result in excessive cell senescence or apoptosis. Impaired proteasome degradation of immune system regulators like IκB can result in immune deficiency. Protein aggregation can also impair chaperone-mediated autophagy, as in the case of the aggreations of the presynaptic protein α-synuclein into the Lewy bodies of Parkinson's Disease [SCIENCE; Cuervo,AM; 305:1292-1295 (2004)].
With aging, lysosomes of postmitotic cells increasingly become bloated with aggregates of oxidized, glycated, cross-linked proteins which are resistant to enzymatic degradation — material called lipofuscin. (When lysosomes become bloated with similar material due to disease processes, it is called ceroid.)
The "error catastrophe" theory of aging proposed that accumulating damage to synthesized proteins resulted in damage to the machinery of synthesis itself, leading to an escalating viscious circle of malfunctioning cellular components. But the rate of both protein synthesis and protein degradation declines with age, and the inability to eliminate damaged macromolecules may be more catastropic than the synthesis of new defective ones.
Ubiquitin levels and protease activity are increased in conditions of stress. Life extension associated with stress response may stimulate DNA repair or anti-oxidant enzyme production, but it can also be a form of hibernation & reduced functionality insofar as shielded proteins are less capable of performing their normal functions.
The production of heat-shock proteins (HSPs — cellular protection from thermal and other stresses) can be increased by a transient elevation of temperatures that could ordinarily kill a cell. Increase longevity and robustness resulting from sublethal stress (hormesis) has been demonstrated in fruit flies and nematodes. The magnitude of induction of heat-shock proteins (particularly the Hsp70 family — which are approximately 70 kilodaltons in size) is significantly reduced with aging [EXPERIENTIA 50:1092-1098 (1994)]. The reported incidence of heat stroke among those 65 years of age or older is ten times that of younger persons.
Although originally discovered in Drosophila (fruit flies) in response to heat, HSPs are now known to also function against other cell stresses such as irradiation, metal poisons and oxidation (even exercise). HSPs enhance cytokine signalling and antigen presentation to lymphocytes [ANNALS OF THE NEW YORK ACADEMY OF SCIENCES; Moseley,PL; 856:206-213 (1998)]. Many cancer cells over-express HSPs, enhancing their survival. HSPs are of remarkably similar structure in nearly all cells, including those from bacteria, plants and mammals. Birds, however, have a unique heat shock transcription factor that is induced in cell proliferation [THE FASEB JOURNAL; Pirkkala,L; 15(7):1118-1131 (2001)] — suggestive of the possibility that HSP could be another factor underlying the exceptional longevity of birds.
Many HSPs are constitutively expressed (rather than expressed by induction), such as the members of the ATP-driven Hsp70 family that reside by ribosomes to assist in folding of newly formed proteins. Some HSPs are true "molecular chaperones" that assist other proteins in transit across intracellular membranes. Some HSPs may protect telomere proteins or telomerase. Some HSPs evidently act by binding to incompletely folded metabolic proteins, protecting them in an inactive state until the traumatic stress has passed. Elderly transgenic mice that overexpress Hsp70 show a recovery of muscles from exercise comparable to that seen in young mice [THE FASEB JOURNAL; Pirkkala,L; 18(2):355-367 (2004)]
Increased expression of the small HSP proteins in the motoneurons of Drosophila has increased lifespan by 15% when expressed in the cytoplasm (Hsp23) and 30% when expressed in mitochondria (Hsp22). Despite the fact that expression was limited to motoneurons, the flies showed an increase in resistance to oxidative injuries by paraquat of up to 35% [THE FASEB JOURNAL; Morrow,G; 18(3):598-599 (2004)]. The longevity effect of small HSPs may be due to their ability to prevent toxic aggregations of proteins. Heat-shock protein response is reduced in aging cells and is elevated in the cells of CRAN (Caloric Restriction with Adequate Nutrition) organisms. Quercetin promotes apoptosis in cancer cells (among other cells) by inhibiting the synthesis of heat shock protein HSP70 [CANCER RESEARCH; Wei,Y; 54:4952-4957 (1994)]. If stress resistance is primarily due to HSPs, it is noteworthy that stress resistance of fibroblasts from 8 mammalian species correlates linearly with species lifespan for a variety of stresses [FREE RADICAL BIOLOGY & MEDICINE; Kapahi,P; 26(516):495-500 (1999)].
Cell structure and metabolism operates under the direction of genes, which are located in the DNA (DeoxyriboNucleic Acid) of the chromosomes of the animal cell nucleus. DNA coding is determined by 4 nucleic acid bases: Adenine, Thymine, Cytosine and Guanine. Adenine and Guanine are known as purines, whereas Thymine and Cytosine are pyrimidines. RNA (RiboNucleic Acid) also contains 4 nucleic acid bases, but differs from DNA by using the base Uracil in the place of Thymine (Uracil is also a pyrimidine).
The nucleic acid bases combine with either a ribose or
deoxyribose sugar molecule to form nucleosides: Adenosine,
Cytidine, Guanosine and Uridine in RNA — or
Deoxyadenosine, Deoxycytidine, Deoxyguanosine
and Deoxythymidine in DNA. Addition of phosphate groups to nucleosides
results in nucleotide phosphates, also called nucleotides.
The nucleotides in RNA are Adenylate, Cytidylate, Guanylate
and Uridylate. Adenosine TriPhosphate (ATP) and deoxyAdenosine
TriPhosphate (dATP) are nucleotides.
| PYRIMIDINES | PURINES |
|---|---|
![[ PYRIMIDINES ]](pyrimidines.gif)
|
![[ PURINES ]](purines.gif)
|
DNA serves as the template (model for copying) for production of both DNA
& RNA. DNA replication (and some DNA repair) is catalyzed by the enzymes
known as DNA polymerases. The production of messenger RNA (mRNA) using
DNA as a template is known as transcription, and is catalyzed by
RNA polymerase II enzyme. Once produced, mRNA leaves the
nucleus for translation of the mRNA code into protein on the ribosomes.
| PURINE/PYRIMIDINE BASE-PAIRINGS | BASE-PAIRINGS IN DNA STRANDS | |
|---|---|---|
![[PURINE/PYRIMIDINE BASE-PAIRINGS]](bases1.gif)
|
![[BASE-PAIRINGS IN DNA STRANDS]](bases2.gif)
|
In normal DNA, the bases Adenine and Thymine are always paired (connected by 2 hydrogen bonds) and the bases Cytosine and Guanine are always paired (connected by 3 hydrogen bonds). A specific sequence of 3 bases in DNA will cause the selection of a single amino acid for protein synthesis. For example, GCA (Guanine, Cytosine, Adenine) will select the amino acid Alanine in the synthesis of structural proteins and enzymes. A gene is a hereditary unit composed of a sequence of DNA bases that will code for a sequence of amino acids that form a peptide or protein.
The DNA bases are connected to sugar molecules (deoxyribose) and the sugar molecules are linked together by phosphate molecules. More precisely, an ester bond (oxygen bond) connects the 5' carbon of one deoxyribose and another ester bond connects the 3' carbon to another deoxyribose, and both ester bonds are connected to a phosphate, forming a 5',3'−phosphodiester bond. The phosphate-linked sugars connected to the bases form a single strand of DNA, which pairs with an antiparallel strand of 3',5'−phosphodiester bonds to form the DNA double-helix.
| Single Strand of DNA |
|---|
![[Single DNA strand]](DNAstrnd.gif)
|
Animal genetic material in the cell nucleus exists as a complex known as chromatin — which consists of DNA, five histone proteins and some non-histone proteins. Histone protein is not coagulated by heat and is composed of a high proportion of the basic amino acids lysine & arginine, which are positively-charged at physiological pH. Because DNA is negatively-charged (due to phosphate groups), the positively-charged histones readily bind to DNA.
Four of the histones (H2A, H2B, H3 and H4) compact DNA about six-fold
into bead-like nucleosomes. A fifth histone (H1) binds to the DNA
between nucleosomes, causing a second-order compacting of the "string"
— compacting the chromatin another six-fold. Non-histone proteins aid
an even higher level of looping & coiling. With age, compacting of
chromatin increases, probably due to increasing covalent linking
between DNA and the chromosomal proteins. Because compacting helps determine
which genes are expressed and which genes are not, the increased
compacting of aging probably means a decline in gene expression. A 50%
reduction in chromatin-associated RNA polymerase II activity has been
demonstrated in the brains of old rats [MUTATION RESEARCH 275:317-329
(1992)].
| BEAD-LIKE NUCLEOSOMES | H1-COMPACTED NUCLEOSOMES | |
|---|---|---|
|
|
Of the approximately 30,000 genes in the human genome, it is estimated that only 2% of these are different from those of a chimpanzee, which has half the estimated maximum lifespan of a human. The longevity difference could be due to as few as a hundred genes or less. Also of note is the fact that identical twins tend to die within 3 years of each other, whereas fraternal twins tend to die within 6 years. Aging theories associated with DNA include programmed aging (or programmed aging-resistance) and theories that link aging with DNA damage/mutation or DNA repair capability.
"Wear&tear" of DNA can take two forms: mutation and DNA damage. An analogy illustrates the difference: the word STOP can be mutated to the word STEP by the substitution of another letter, whereas if the letter "O" is lost or altered, damage occurs, resulting in the non-word ST#P. Substitution of a Thymine for an Adenine would be a mutation, whereas loss of an Adenine or methylation of a Guanine would be damage. The phenomena are not independent, however, because methylated Guanine is known to be mutagenic. Of chemicals known to be mutagenic in bacteria 85% are carcinogenic (cancer-causing) in animals &mdas the basis of the Ames Test for carcinogenicity.
DNA damage tends to interfere with gene expression by preventing transcription of RNA from DNA, whereas mutation usually results in transcription that usually produces proteins with diminished or altered functionality. Mutations that are not lethal to a cell are more likely to be perpetuated in dividing cells. DNA damage rather than DNA mutation is posited as a cause of aging.
There are more than 200,000 DNA damage events per mammalian cell per day due to oxidation, hydrolysis, alkylation, radiation or toxic chemicals. Removal of purine or pyrimidine bases from DNA (depurination and depyrimidation ) is often caused by hydrolysis or thermal disruption. The location on DNA where a depurination or depyrimidation has occurred is called an AP site (APurinic or APyrimidinic site). If AP sites are unrepaired they decay to single-strand breaks. Pyrimidine dimers (usually cross-linking of two adjacent thymine bases) frequently are produced by ultraviolet light.
Types and frequency of DNA damage can be roughly illustrated by the following table and representative pictures:
![[DEPICTIONS OF DNA DAMAGE]](break.gif)
TYPE OF DAMAGE | events/cell/day | % of total daily damage |
|---|---|---|
| Single-strand break | 120,000 | 50.9 |
| N7-MethylGuanine | 84,000 | 35.6 |
| Depurination | 24,000 | 10.2 |
| O6-MethylGuanine | 3,120 | 1.3 |
| Oxidized DNA | 2,880 | 1.2 |
| Depyrimidation | 1,320 | 0.5 |
| Cytosine deamination | 360 | 0.2 |
| Double-strand breaks | 9 | 0.01 |
| Interstrand cross-links | 8 | 0.01 |
| Illustrated Summary of Types of DNA Damage |
|---|
![[DNA Damage Illustration]](DNAdamage.gif)
|
| 8−OHdG from Guanosine with Hydroxyl | 8−oxoG |
|---|---|
![[ 8−HydroxdeoxyGuanosine ]](OH_dG.gif)
|
![[8−oxo−7,6−dihydroGuanine ]](oxoG.gif)
|
More than 20 types of oxidative damage to nucleosides have been documented [THE FASEB JOURNAL; Cooke,MS; 17(10):1195-1214 (2003)]. The most frequent oxidative damage to DNA is believed to be the 8−hydroxylation/oxidation of the guanine base to 8−hydroxydeoxyguanosine (8−OHdG), a molecule which is equivalent to 8−oxo−7,8−dihydroguanine (8−oxoG) because the hydroxyl hydrogen can easily move to the 7−position leaving a double-bonded oxygen at the 8−position (a resonance form of the two structures). Singlet oxygen reacts with DNA to produce 8−OHdG/8−oxoG [JOURNAL OF BIOLOGICAL CHEMISTRY; Ravanat,J; 275(51):40601-50604 (2000)]. 8−OHdG/8−oxoG is the most commonly studied biomarker of DNA oxidation [MUTATION RESEARCH 424:51-58 (1999)] and is believed to constitute 5% of all oxidative DNA damage [MUTATION RESEARCH; Dizdaroglu;M; 275(2-6):331-342 (1992)]. 8−OHdG is mutagenic because it inhibits methylation and because it can be paired with adenosine rather than cytosine during DNA replication leading to GC-to-AT conversion (the most frequent kind of spontaneous mutation). Levels of 8−OHdG are inversely related to lifespan in mammals [FREE RADICAL BIOLOGY & MEDICINE; Foksinski,M; 37(9):1449-1454 (2004)], and increase with age — but less so in animals subjected to Caloric Restriction with Adequate Nutrition (CRAN) [FREE RADICAL BIOLOGY & MEDICINE 32(9):882-889 (2002)]. Mitochondrial DNA rather than nuclear DNA is the primary site of damage [THE FASEB JOURNAL; Barja,G; 14(2):312-318 (2000)]. In Western countries, females live about 10% longer than males, and males have 4 times as much oxidative DNA damage as females, presumably because females have more MnSOD and glutathione peroxidase [FREE RADICAL BIOLOGY & MEDICINE; Borras,C; 34(5):546-552 (2003)]. Levels of 8−OHdG are 18 times higher than normal in intact DNA from the cerebrospinal fluid of Alzheimer's Disease patients [ARCHIVES OF NEUROLOGY 58:392-396 (2001)]. Nuclear DNA in the brain tissue of old mice accumulates 8−OHdG/8−oxoG at nearly four times the rate of young mice [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Hamilton,ML; 98(18):10469-10474 (2001)]. Repair of 8−OHdG/8−oxoG has been shown to decline significantly with age in humans [JOURNAL OF RADIATION RESEARCH; Chen,S; 44(1):31-35 (2003)]. Cigarette smoking, age, and unhealthy diet correlate directly with not only urinary 8−OHdG/8−oxoG, but urinary N7-MethylGuanine [CANCER SCIENCE; Tamae,K; 100(4):715-721 (2009)].
The most active DNA repair enzymes, excision repair enzymes, all operate on the basis of damage or mutilation occurring to only one of the two strands of the DNA double-helix such that the undamaged strand can be used as a template to repair the damaged strand. The damaged area of the injured strand is cut-away (excised) by a nuclease (or glycosylase) enzyme, and a new strand (or a single nucleotide) is constructed. Even the simplest repair usually involves a team of enzymes.
Glycosylase (glycosidase) enzymes remove individually damaged nucleic acid bases (purines or pyrimidines) from the deoxyribose sugars to which they are attached. Glycosylases remove bases that have been oxidized or alkylated and also remove uracil from nDNA. The MYH glycosylase removes adenine that has been incorrectly incorporated opposite 8−OHdG/8−oxoG. Endonuclease enzymes cleave the phosphodiester bonds to remove the sugar residues (which may or may not still be connected to a base). There are at least fifteen DNA polymerase enzymes which function in DNA repair to replace excised strands of DNA. DNA ligase enzymes seal the strand by reforming the phosphodiester bonds. If a long section of strand needs to be replaced, helicase enzymes may be required to unwind the DNA before the injured section is excised — and rewind afterwards. Very long sections may even require topoisomerase enzymes to unwind and rewind supercoils. Additional enzymes are often required to recognize damage and recruit other enzymes into repair.
There are three general categories of excision−repair enzymes:
(1) Base Excision Repair (BER, which repair/replace a single damaged
nucleic acid base) (2) Nucleotide Excision Repair (NER, for repairing
DNA strand damage ranging from 2−30 bases in length) and (3) MisMatch Repair
(MMR, for repairing mispaired nucleic acid bases).
|
|
Base Excision Repair (BER) primarily repairs damage due to hydrolysis, alkylation (usually methylation) or oxidation of single nucleic acid bases. Alteration of a single base may not impede transcription and can often lead to miscoding and thus to mutation. BER begins with recognition and removal of a damaged nucleic acid base by one of many possible glycosylase enzymes, each of which specializes in recognition of a particular type of base damage [BIOCHEMICAL JOURNAL; Krokan,HE; 325(Pt 1):1-16 (1997)]. BER has two subpathways, known as short-patch BER and long-patch BER. Roughly 80−90% of BER is by the short-patch pathway, which requires only 3 enzymes: a glycolsylase, an endonuclease and a polymerase. Bifunctional glycosylases not only cleave the bond between the damaged base and the sugar, but cleave the backbone with AP lyase activity. But for obstinate base modification that cannot be fixed by the short-patch pathway, the long-patch pathway strips-away 2−10 nucleotides, including the damaged base. A larger number of proteins participate in the long-patch pathway, such as PCNA, RFC, FEN1 and probably WRN. PCNA, RFC and a polymerase create a "flap" of nucleotides that are removed by FEN1 (Flap ENdonuclease−1);
Deamination of cytosine by hydrolysis is an example of DNA damage repaired by short-patch BER. With the removal of the (−NH2) group, cytosine becomes uracil, which is recognized by DNA repair enzymes as being an abnormal base in DNA. (The fact that cytosine deaminates so easily to uracil probably explains why thymine rather than uracil is normally present in DNA — it is easier to detect a base not normally present). The repair enzyme uracil-DNA glycosylase removes the uracil and then an AP endonuclease cleaves the phosphodiester bonds, just as it would in the repair of any depurination or depyrimidation. DNA polymerase ß is a specialized DNA polymerase that is used for attaching the new base in BER, not for DNA replication. BER repair capabilities dependent upon DNA polymerase ß have been shown to decline with age in mice, which may underly increased vulnerability to cancer or even aging itself [MUTATION RESEARCH 500:135-145 (2002)]. Normal BER forms transient single-strand breaks, so it is understandable that BER enzymes play an important role in single-strand break repair. There are no known diseases associated with inherited defects of short-patch BER enzymes. Individual glycosylase defects are not harmful because there are so many glycosylases which can perform the same functions, whereas defects in the other short-patch BER enzymes are fatal to embryos.
One might imagine that increased expression of BER enzymes would improve DNA integrity,
but the opposite is true. Increased glycosylase expression increases DNA strand
breaks [MOLECULAR CANCER THERAPIES; Rinne,M; 3(8):955-967 (2004)] as
does increased DNA polymerase ß
expression [NUCLEIC ACIDS RESEARCH; Canitrot,Y; 32(17):5104-5112 (2004)].
Increased AP nuclease expression can increase genetic
instability [NUCLEIC ACIDS RESEARCH; Sossou,M; 33(1):298-306 (2005)]. Insofar as
the stages of DNA repair involve creation of AP sites and clipping of DNA strands, these
results should not be surprising. Enhanced DNA repair would require co-ordinated
increase in many enzymes.
|
Nucleotide Excision Repair (NER) repairs damage affecting more than one
nucleic acid base, defects which distort the DNA helix and can be exemplified by the
repair of cross-links between purines & the deoxyribose-phosphate backbone due to the
hydroxyl radical and by pyrimidine dimers (CPDs, Cyclobutane Pyrimidine Dimers,
two covalently-bonded adjacent pyrimidines, usually thymine dimers) caused by
ultraviolet light. Thymine-cytosine and cytosine-cytosine are the most mutagenic
CPDs [JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY; Marrot,L;
58(5 Suppl 2):S139-S148 (2008)]. Less frequently than CPDs, ultraviolet
light also induces 6−4 pyrimidine-pyrimidone photoproducts
(6−4 PPs). CPDs and 6−4 PPs lead to
apoptosis [BMC CANCER; Lo,H; 5:135 (2005)] or double-strand
breaks [THE EMBO JOURNAL; Garinus,GA; 24(22):3952-3962 (2005)]
if not repaired by NER. Carcinogen lesions like those caused by aflatoxin (which forms bulky
DNA adducts) are also corrected by NER. DNA polymerase delta and DNA polymerase epsilon
are the specialized DNA polymerases used in NER. Many steps and more than 20 proteins are
involved in unwinding the DNA, in recognizing the type
of damage to be repaired, etc. NER provides backup to BER when glycosylases are defective in
the nucleus, but NER systems are absent from mammalian mitochondria (which only have BER).
A consequence of the fact that NER is so much more complex than BER is the fact that NER is more
error-prone than BER. A study of seven mammalian species showed a correlation between
both rate and extent of NER after UV exposure and lifespan of the
species [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Hart, RW;
71(6):2169-2173 (1974)].

|
There are two subtypes of NER, distinguished by how damage is recognized: (1) Global-Genome Repair (GGR, recognizes damage throughout the genome) and (2) Transcription-Coupled Repair (TCR, recognizes damage by stalled transcription). The slower GGR NER (like all the DNA repair mechanisms other than TCR) gradually covers the whole exposed genome. GGR recognizes strand defects with XP protein — so-named because defects in these helicase (DNA helix unwinding) proteins (identified alphabetically from XPA to XPG) lead to the disease known as Xeroderma Pigmentosum. The XP helicase unwinds DNA in the area of the DNA damage so that other NER enzymes can make the repair. XPB & XPD are subunits of Transcription Factor IIH (TFIIH), which functions in normal transcription as well as in NER. XPF−ERCC1 is an endonuclease wherein XPF is the catalytic component and ERCC1 is required for DNA binding. TCR NER is a preferential NER pathway focused on genes that are being transcribed. TCR ensures that DNA that is actively being transcribed is given the highest priority for repair. Typically a TCR enzyme detects a stalled RNA polymerase which is unable to proceed because of the DNA damage. The detection proteins are called CS proteins because when they are defective the result is a disease known as Cockayne Syndrome. CS proteins aid in displacement of the stalled RNA polymerase to allow NER enzymes to access the damaged DNA. Then the XP helicase does the unwinding and TCR NER then proceeds much as it would for global NER. The tumor-suppressor protein BRCA1 (which is often defective in breast cancer) is essential for TCR associated with oxidative DNA damage (but not with TCR associated with ultraviolet light damage) [SCIENCE; Gowen,LC; 281:1009-1012 (1998)]. Defects in GGR lead to cancer, whereas defects in TCR more readily lead to apoptosis ("accelerated aging") [NATURE REVIEWS; Ljungman,M; 4(9):727-737 (2004)]. Nonproliferative cells do not exhibit GGR, only exhibiting DNA repair on transcribed genes [MOLECULAR AND CELLULAR BIOLOGY; Nouspikel,T; 20(5):1562-1570 (2000) and MUTAGENESIS; Bielas,JH; 21(1):49-53 (2006)].
MisMatch Repair (MMR) corrects errors made during DNA copying, such as the mispairing of an adenosine base with a guanosine. MMR can correct A−C & T−C mismatches more efficiently than G−A & T−C mismatches, and does a very poor job of correcting C−C mismatches [BIOCHEMICAL JOURNAL; Marra,J; 338(Pt 1):1-13 (1999)]. But how do the recognition enzymes know which is the correct base, the adenosine or the guanosine? For bacteria, the answer is known: when DNA is freshly synthesized the parental strand has methyl groups attached to certain adenosine residues, whereas the newly synthesized strand will be unmethylated for some time after replication. Prior to methylation of the new strand the detection enzymes can look for errors. DNA methylation is apparently not used for error-detection in multi-cellular organisms, however, and the means of mismatch detection is still unknown. Mismatch repair differs from BER only in the first glycosylase, which recognizes and removes mispaired bases — in contrast to BER which recognizes and removes defective bases. Removing the mispaired base leaves an AP site which can then be repaired by the subsequent BER enzymes. Failures in MMR result in mutations, whereas failures in BER result in DNA damage (including mutations). Defects in MMR operation result in mutation rates 100−fold greater than seen in normal cells, most often in microsatellite sequences. MMR corrects not only single base mispairs, but Insertion/Deletion Loops (IDLs) that result from strand misalignments, which can produce frameshift mutations (disrupted triplet codon reading due to insertion or deletion of base pairs that is not a multiple of 3). As well, MMR plays a significant role in protecting against incorporation of 8−OHdG/8−oxoG into DNA [MOLECULAR AND CELLULAR BIOLOGY; Russo,MT; 24(1):465-474 (2004)]. Hereditery NonPolyposis Colon Cancer (HNPCC) is often caused by defective Msh2 protein, which normally functions to recognize mispaired bases and to signal MMR [JOURNAL OF BIOLOGICAL CHEMISTRY; Mazurek,AM; 277(10):8260-8266 (2002)]. MMR also protects against cancer by inhibition of ALT (Alternative Lengthening of Telomeres) [CANCER RESEARCH; Bechter,OE; 64(10):344-3451 (2004)].
DNA repair enzymes exist for double-stand breaks and for guanine methylation, neither of which involve excision of single DNA strands. Methylation of cytosine is a normal means by which cells prevent gene expression. But methylation of guanine is DNA damage, and deamination of a methylated cytosine results in thymine — a mutation. A "suicidal" methyl transferase enzyme can repair O6−methylguanine by transferring the methyl group to its own cysteine. The DNA repair enzyme O6−MethylGuanine-DNA MethylTransferase (MGMT) is frequently repressed by hypermethylation in colon cancer, which thereby allows alkylating agents to cause the G:C-to-A:T conversions which are behind the K−ras mutation seen in about half of colorectal carcinomas. MGMT can remove not only methyl groups from guanine, but chloroethyl and benzyl groups. Because MGMT corrects the nucleotide without removal, it is said to do repair by Direct Reversal (DR).
Double-Strand Breaks (DSBs) normally occur during V(D)J recombination and meiosis where genetic recombination can be beneficial. Damaging DSBs are usually due to ionizing radiation or very high doses of alkylating carcinogens such as nitrogen mustards. When the damage is not so severe Single-Strand Breaks (SSBs) may result. Even with ionizing radiation, double-strand breaks are only produced with about a tenth the frequency of single strand breaks. Although double-strand breaks are rare, they are difficult to repair and can be very injurious for somatic cells that undergo mitosis. (The Deinococcus radiodurans bacterium that lives in nuclear reactors repairs double-strand damage very efficiently.) DSBs are repaired by (1) Non-Homologous End-Joining (NHEJ) or (2) Homologous Recombination (HR). NHEJ is the simplest and most common means of DSB repair, but it is the least accurate. For NHEJ the two broken ends are rejoined without regard to deletions or rearrangements. By contrast, HR exactly reconstitutes the original sequence of genes because the sister chromatid (during mitosis) or homologous chromosome (during meiosis) is used. But HR is limited by the fact that it can only operate during meiosis or late mitosis. NHEJ is the only repair mechanism available for non-mitotic cells, whereas for mitotic cells NHEJ operates in the G0, G1 and early S phase of the cell cycle, whereas HR operates in late S phase and G2.
DSBs are recognized by the MRN complex (composed of Mrell, Rad50 and Nbs1
proteins), which unwinds the DNA ends and recruits ATM protein to the
site of the break [SCIENCE; Lee,J; 308:551-554 (2005)]. ATM phosphorylates
H2AX histone (which recruits DNA repair proteins [SCIENCE; Celeste,A;
296:992-997 (2005)]) and p53 protein (which blocks progression
through the cell
cycle leading to DNA repair or apoptosis — if DNA damage is too great
for available DNA-repair resources). ATM is responsible for phosphorylation of
Rad51 protein required for
HR [JOURNAL OF BIOLOGICAL CHEMISTRY; Chen,G; 274(18):12748-12752 (1999)].
ATM regulates not only HR, but a more precise form of
NHEJ [CANCER RESEARCH; Wang,H; 66(3):1391-1400 (2006)].
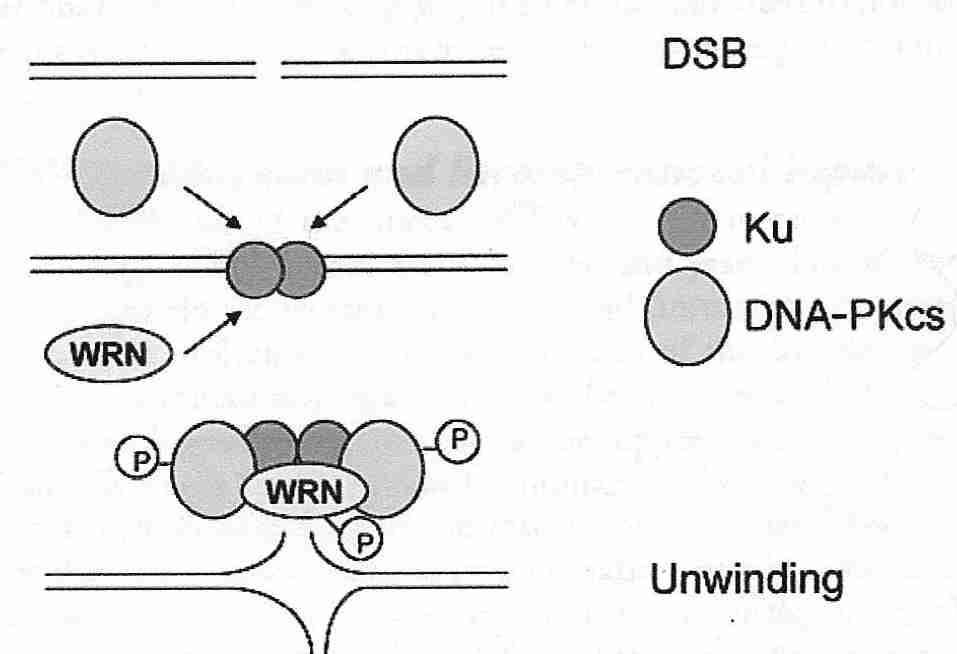
|
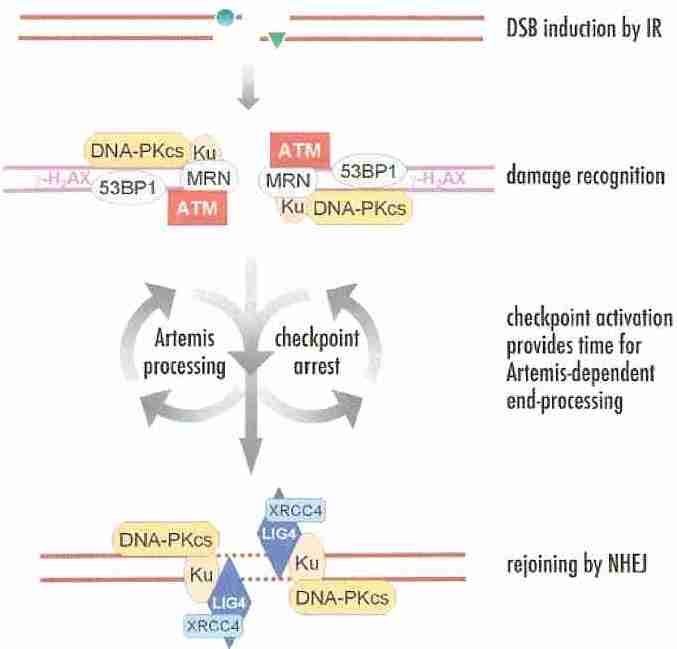
|
DSB repair is even more error-prone than NER, especially in the case of NHEJ. The WRN protein, which is defective in Werner's syndrome, operates in DSB repair by both NHEJ and HR [NUCLEIC ACIDS RESEARCH; Brosh,RM; 35(22):7527-7544(2007)]. WRN protein minimizes nucleotide loss during NHEJ [CANCER RESEARCH; Oshima,J; 62(2):547-551 (2002)]. Cells lacking WRN may have such inefficient HR that they are dependent on forms of NHEJ for DSB repair [GENES & DEVELOPMENT; Prince,PR; 15(8):933-938 (2001)]. The Ku protein heterodimer (Ku70/Ku86) initiates NHEJ by binding to broken DNA ends and bringing them together. (Ku86 (mice?) is sometimes called Ku80, but is actually 83 kiloDaltons.) Ku heterodimers are so plentiful in mammalian nuclei that any DSB is likely to occur within five molecular diameters of a Ku dimer. Both the Ku heterodimer & the DNA-dependent Protein Kinase complex (DNA−PKc) bind to WRN protein and regulate WRN activity [JOURNAL OF BIOLOGICAL CHEMISTRY; Karmakar,P; 277(21):18291-18302 (2002)]. WRN unwinds the DNA strands and then Ku attachment to WRN strongly stimulates endonuclease activity in preparation for ligation [CARCINOGENESIS; Opresko,PL; 24(5):791-802 (2003)]. Overexpression of Ku in normal yeast reduces gross chromosomal rearrangements, but Ku overexpression increases gross chromosomal rearrangement in strains having a defective WRN homolog [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Banerjee,S; 103(6):1816-1821 (2006)]. Ku86 and DNA−PKc are also important for telomere maintenance [THE JOURNAL OF CELL BIOLOGY; Espejel,S; 167(4):627-638 (2004)]. The nuclease Artemis processes damaged DNA ends prior to ligation (rejoining) [CELL CYCLE; Jeggo,PA; 4(3):359-362 (2005)]. The tumor-suppressor protein BRCA1 has also been shown to play a critical role in NHEJ [HUMAN MOLECULAR GENETICS; Deng,C; 12(1):R113-R123 (2003)]. Although NHEJ is "molecular guesswork" and very error prone, it is often effective because most of the genome is composed of "junk DNA". One or more alternate forms of NHEJ exist, and these alternate NHEJ forms are even more error-prone and mutagenic than primary NHEJ [PLoS GENETICS; Bennardo,N;4(6):1-10(2008)]. Ku protein has a much higher affinity for DNA ends than PARP−1 does, but in the absence of Ku PARP−1 will bind to DNA ends to provide a more error-prone "backup" form of NHEJ [NUCLEIC ACIDS RESEARCH; Wang,M; 34(21):6170-6182 (2006)].
Homologous recombination (HR, also called REcombinational Repair,
RER), a more
accurate but less frequently-used means of DSB repair, is the dominant method used in
the late S and G2 phases of the cell-cycle, after a sister chromatid has
been created. Although HR is primarily restricted to repair of DSBs in proliferating
cells, even in proliferating cells 75% of DSBs are repaired by NHEJ [DNA REPAIR;
Mao,Z; 7(10):1765-1771 (2008)]. ATR (ATM and Rad3-related) checkpoint
protein facilitates HR [CANCER RESEARCH; Wang,H; 64(19):7139-7143 (2004)] and senses stalled
DNA replication forks [DNA REPAIR; Paulsen,RD; 6(7):953-966 (2007)], but ATR does
not facilitate NHEJ [CANCER RESEARCH; Wang,H; 64(19):7139-7143 (2004)]. (ATR is most active in
proliferative tissues.) In one form of HR (synthesis-dependent strand annealing) a single
DNA strand must associate with its complementary strand in a double-stranded DNA
molecule. In a simpler form of HR (single-strand annealing, which requires
fewer proteins) a single strand associates with its complementary single strand.
Homologous pairing of the sister chromatids is often mediated by Rad51
protein, which is normally necessary for cell proliferation and survival. Rad52
protein recognizes the DSB and adheres to the free ends of the break (comparable to
Ku in NHEJ) while Rad51
searches the undamaged sister chromatid for a homologous repair template. The tumor
suppressor protein BRCA2 co-localizes with Rad51 during homologous recombinational
repair, and contributes significantly to its
activity [BREAST CANCER RESEARCH; Orelli,BJ; 3(5):294-298 (2001)]. HR could be
a basis for telomerase-independent telomere lengthening (ALT) in
mammals [THE EMBO JOURNAL; Blasco,MA; 24(6):1095-1103 (2005)].
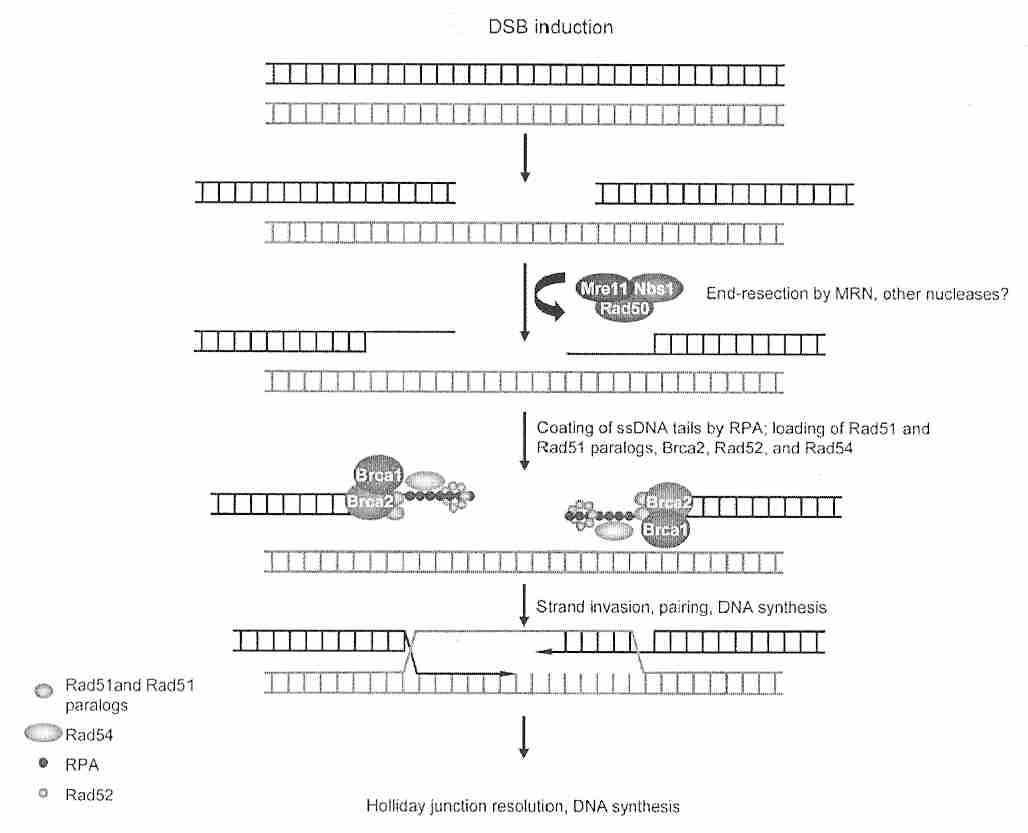
|
For HR during
meiosis there can be a loss of information ("loss of
heterozygosity")
if a gene conversion (replacement of two different
alleles by the
same allele on both chromosomes) occurs.
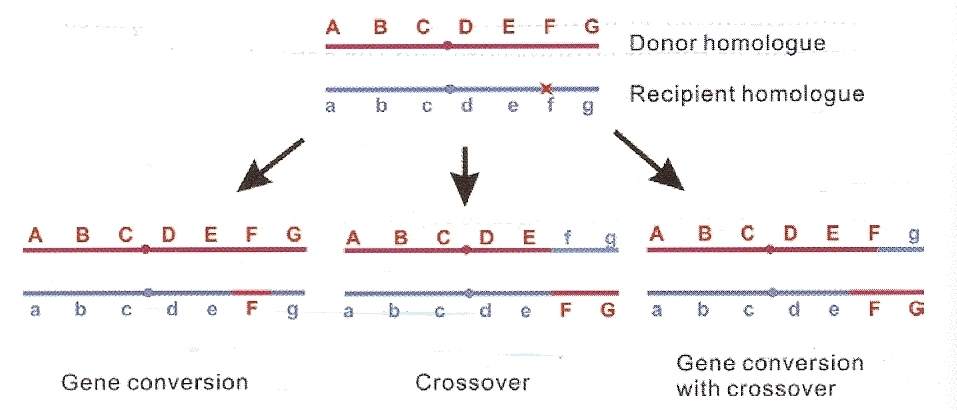
|
TransLesion Synthesis (TLS) uses specialized DNA polymerases to quickly patch damaged strands. Although more error-prone than BER, NER or MMR, TLS may reduce the danger of DSBs [GENES & DEVELOPMENT; Prakash,S; 16(15):1872-1883 (2002)].
The bacterium Deinococcus radiodurans is the most radiation-resistant organism known. Within one day of exposure to radiation inducing hundreds of DSBs the entire genome is usually faithfully restored. The bacterium has a wide range of DNA repair enzymes and a high amount of redundancy in the genes for those enzymes. With 4−10 copies of the entire genome per cell, the polyploid bacterium has access to numerous templates for homologous recombination [SCIENCE; White,O; 286:1571-1577 (1999)].
Single-Strand Breaks (SSBs) must be repaired quickly to prevent them from becoming DSBs. An NAD-dependent enzyme named Poly(ADP-Ribose) Polymerase−1 (PARP−1) binds to SSBs and recruits XRCC1 protein, which provides the scaffold necessary for DNA polymerase β to fill the gap [CELL RESEARCH; Horton,JK; 18(1):48-63 (2008)]. In response to single-strand DNA damage due to alkylating agents, oxidants or ionizing radiation, levels of PARP−1 can increase several hundred-fold. Maximum lifespan in mammalian species correlates with PARP activity [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Grube,K; 89(24):11759-11763 (1992)]. PARP−1 modifies histones, transcription factors and other nuclear regulatory proteins by addition of many ADP-ribose molecules to the glutamic acid residues — increasing negative charge and thereby causing proteins to unfold or become more open. PARP−1 addition of long ADP-ribose tails to histones can quickly decondense chromatin to enable rapid transcription [SCIENCE; Pirrotta,V; 299:528-529 (2003)] and access for DNA repair [SCIENCE; Tulin,A; 299:560-562 (2003)]. The ADP-riboses come from removing nicotinamide from NAD+ (Nicotinamide Adenine Dinucleotide). Poly (ADP-ribosyl)ation is a transient protein modification that is rapidly reversed by poly (ADP-ribose) glycohyrolase. In addition to SSBs, PARP−1 functions in BER and NHEJ to detect DNA damage and recruit DNA repair enzymes [JOURNAL OF BIOLOGICAL CHEMISTRY; Schreiber,V; 23028-23036 (2002) and JOURNAL OF BIOLOGICAL CHEMISTRY; Audebert,M; 279(53):55117-55126 (2004)], but may require the WRN protein to do so [MOLECULAR AND CELLULAR BIOLOGY; 23(23):8601-8613 (2003)]. PARP−1 and WRN seem to interact in NHEJ as well as BER [NUCLEIC ACIDS RESEARCH; Beneke,S; 35(22):7456-7465 (2007)].
Some protection against DNA damage is provided by gene redundancy. But the only structural genes known to be present in multiple copies are those coding for ribosomal RNA (rRNA), transfer RNA (tRNA) and histones. Apoptosis (cell suicide) is the most effective defense against DNA damage & mutation when DNA repair enzymes are inadequate to fix the damage. But p53 induction following UV irradiation declines with age, as do levels of DNA repair proteins [THE FASEB JOURNAL; Goukassian,D; 14(10):1325-1334 (2000)]. The resultant decline of DNA repair associated with decreased apoptosis for DNA damage can contribute to cancer, and probably to aging.
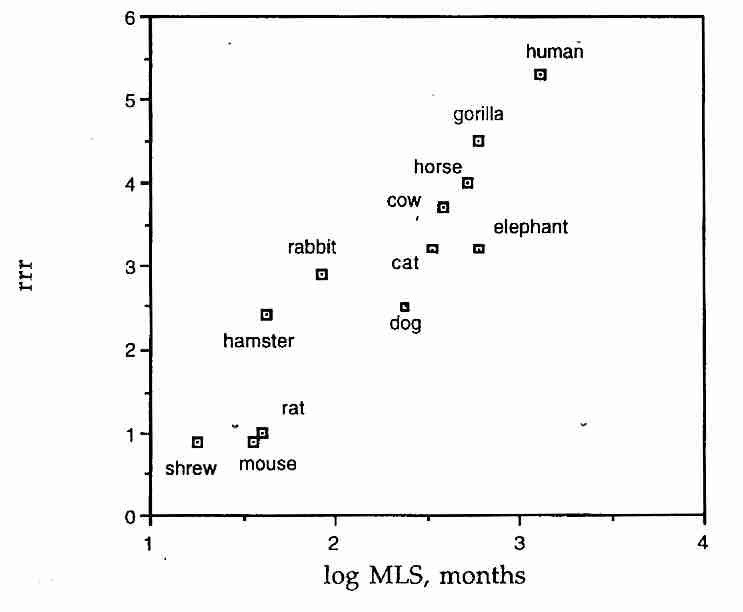
|
The amount of oxidative DNA damage in neurons is likely to be many times greater than in most other cells. The human brain accounts for only 2% of total body weight, but 20% of resting oxygen consumption due to the high metabolic demand required to maintain membrane ion potentials. Neurons transcribe about 2−4 times as much DNA as do cells from kidney, liver or spleen. Yet neurons are non-dividing and must last a lifetime.
A profiling of gene expression for stem cells shows enriched expression of DNA repair genes [SCIENCE; Ramalho-Santos,M; 298:597-600 (2002)].
Even a "wear&tear" theory like DNA damage is subject to a programmed aging interpretation. In general, DNA repair tends to lag behind DNA damage to a greater extent in short-lived species, and the amount of lag can constitute the degree of "programmed aging". A study which correlated maximum lifespan in a variety of mammalian species found a six-fold difference in the nuclear DNA-repair activity of mice and men. A graph of DNA-repair activity standardized on the rat ("rat-relative repair", where rat DNA-repair activity equals 1.0), showed a direct correlation between rat-relative DNA repair and maximum lifespan for the species [MECHANISMS OF AGING AND DEVELOPMENT; Cortopassi, GA; 91:211-218 (1996)].
Many of the nuclear DNA (nDNA) repair enzymes discussed above are the same as
or similar to the enzymes that repair mitochondrial DNA (mtDNA). The main deficiency
in mtDNA repair is the absence of NER enzymes/proteins, but the presence of multiple
copies of mtDNA in each mitochondrion compensates somewhat. The multiple genomes also
makes HR (homologous recombination) more feasible for mtDNA DSB repair.
|
Because of the rapid turnover of mitochondria in cells, oxidative damage to mitochondrial lipids (membranes) and proteins is normally less of a concern than oxidative damage to mtDNA. But with age, lysosomes become less efficient at removing defective mitochondria. Oxidative damage to cardiolipin in the inner mitochondrial membrane reduces oxidative phosphorylation [GENE; Paradies,G; 286(1):35-41 (2002)], which is probably an important factor in the declining ATP production by mitochondria associated with aging [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Short,KR; 102(15):5618-5623 (2005)]. Moreover, the enzymes responsible for importing DNA repair proteins into the mitochondria become increasingly defective with age (possibly due to oxidative damage) [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Szczesny,B; 100(19):10670-10675 (2003)].
Healthy adults 65−80 years of age have about 25% higher skeletal muscle mtDNA 8−oxodG than adults 20−35 [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Short,KR; 102(15):5618-5623 (2005)]. Oxidative damage to mtDNA leads to mtDNA deletions [EXPERIMENTAL BIOLOGY AND MEDICINE; Wei,Y; 227(9):671-682 (2002)]. Clonal expansion of mtDNA deletions with age may ultimately affect nearly all somatic mtDNA, leading to degenerative disease and the aging phenotype [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Nekhaeva,E; 99(8):5521-5526 (2002)]. Even without oxidative damage mtDNA mutations and deletions promote apoptosis, leading to tissue degeneration and aging [SCIENCE; Kujoth,GC; 309:481-484 (2005)]. One mouse model of "accelerated aging" is based on a mtDNA polymerase knockout that leads to a 3−5 fold increase in point mutations, as well as to increased mtDNA deletions [NATURE; Trifunovic,A; 429:417-423 (2004)].
Animal cells can be classified as germ cells (sperm or egg), stem cells (undifferentiated cells that can differentiate into functioning body cells) and somatic cells (differentiated functioning body cells). Somatic cells are either non-dividing after birth (like neurons or muscle cells) or cells that continue to divide (stem cells and most somatic cells).
One of the most famous experiments in biogerontology was done by Leonard Hayflick. He observed that embryonic fibroblasts (connective tissue cells) in tissue culture would divide about 50 times before they ceased dividing. This 50−division limit (the Hayflick Limit) seemed to be a property of the cell nucleus or DNA.
A human somatic or stem cell has 23 chromosome pairs (46 chromosomes). Because each chromosome has two ends, there will be 92 chromosome ends per cell. At the ends of each chromosome is a long non-functional strand of DNA called a telomere. Telomeres consist of the six-base repeating sequence TTAGGG (2 Thymines, 1 Adenine and 3 Guanines). With each cell division, some of the telomere is lost because DNA polymerase cannot complete the 5'−end and therefore leaves a single-strand 3'−end overhang. But the number of times that most dividing cells can divide is limited by telomere length.
At conception each human telomere is about 10,000 base pairs long (ie, about 1,666 TTAGGG repeats), and the typical chromosome is about 13 thousand times longer (130 million base-pairs). Nine months later, at birth, the average telomere is half as long as it was at conception. Telomeres lose an average of eight TTAGGG subunits per cell division, so half of the telomere length was lost due to the cell divisions of embryonic development. Human telomeres are less than half as long as the telomeres of other primates — and the telomeres of rodents are longer than those of primates [BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS 263:308-314 (1999)]. Telomeres shorten more rapidly in short-lived mammals & birds than in long-lived ones [PROCEEDINGS; BIOLOGICAL SCIENCES; THE ROYAL SOCIETY; Haussmann,MF; 270(1522):1387-1392 (2003)].
For some species there is a correlation between maximum lifespan and the number of fibroblast doublings for that species. Fibroblasts from different species of mammals display a direct relationship between species lifespan and number of populations doublings, from 8-11 in mice to 57-67 in humans [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Rohme,D; 78(8):5009-5013 (1981)]. Among non-mammals, chickens with a 12−year lifespan show 25 doublings and the Galapagos tortoise with a 175−year lifespan shows 130 doublings. These species not only differ in initial telomere length, but in the number of telomeres lost at each cell division. But if maximum lifespan was determined by the Hayflick Limit alone, these species would have a lifespan 2−3 times greater than what is observed. Mice have very long telomeres, but have a very short lifespan — showing that long telomeres need not mean high replicative capacity. Flies & nematodes are comprised entirely of post-mitotic (non-dividing) cells, which means telomeres are of no relevance to lifespan in those species. Large animals tend to require more cell divisions and also live longer, but this does not mean that a large number of divisions causes longevity.
For humans, the length of the remaining telomere is usually an indicator of how many divisions a dividing cell has left. One study found an inverse relationship between telomere length and pulse pressure, indicating a possible direct relationship between vascular aging and telomere length [HYPERTENSION; Jeanclos,E; 36(2):195-200 (2000)]. Higher levels of oxidative stress increase the rate of telomere shortening [TRENDS IN BIOCHEMICAL SCIENCES 27(7):339-344 (2002)]. Once the telomere is gone, functional genetic DNA would be lost with each cell division. Prior to complete erosion of the telomere a signal is sent to p53 protein (possibly by ATM protein) to stop the cell cycle causing the cell to go into a slow-decaying, non-replicative state known as replicative senescence. Telomeres protect chromosomes like the plastic cap that prevents shoe-laces from becoming frayed at the ends. Telomeres have been shown to be seven times more vulnerable to hydroxyl radical oxidation than similar-sized DNA control fragments, indicating that telomeres could sacrificially protect coding DNA from oxidative damage [JOURNAL OF BIOLOGICAL CHEMISTRY; Henle,ES; 274(2):962-971 (1999)].
|
Telomeres are actually a loop-like structure which is associated with an assortment of proteins (the shelterin complex), the most notable of which are the Telomeric Repeat-binding Factors (TRFs). TRF1 regulates telomere length, assisting the telomerase enzyme. TRF2 models the telomere into the T−loop structure. TRF2 may be protecting the single-stranded 3'−end overhang from degradation, and by binding to ATM prevents the ATM-dependent DNA damage response [THE EMBO JOURNAL; Blasco,MA; 24(6):1095-1103 (2005)]. Loss of TRF2 from telomeres directly signals apoptosis [SCIENCE; Karlseder,J; 283:1321-1325 (1999)]. TRF2 stimulates the helicase activity of both WRN (of Werner's Syndrome) and BLM (of Bloom Syndrome), which may play a role in telomere maintenance [JOURNAL OF BIOLOGICAL CHEMISTRY; Opresko,PL; 277(43):41110-41119 (2002)]. Ku proteins (normally active in double-strand break repair) prevent aberrant telomere-telomere fusions. Tankyrase is a PARP — Poly (Adenosine diphosphate-Ribose) Polymerase — which can ADP-ribosylate TRF1, thereby removing it from DNA and allowing telomerase lengthening of the telomere [SCIENCE; Pirrotta,V; 299:528-529 (2003)]. TRF2 is regulated by PARP−2 [MOLECULAR AND CELLULAR BIOLOGY; Dantzer,F; 24(4):1595-1607 (2004)].
Germ cells, stem cells and "immortalized" cancer cells contain an enzyme called telomerase that replaces lost telomeres, thus preventing them from experiencing a Hayflick Limit. Telomerase is a reverse transcriptase, meaning an enzyme that makes DNA from an RNA template (the reverse of normal transcription which uses DNA as the template for making RNA). In human germ cells or 85% of cancer cells human TElomerase Reverse Transcriptase (hTERT) and an RNA template are sufficient conditions for the creation of new telomeres. Because most cells normally express the RNA template, derepression of hTERT is the critical step for acquiring telomerase activity. Defects in proteins required to maintain telomere function can also lead to chromosome instability and cancer [EXPERIMENTAL GERONTOLOGY 36:1619-1637 (2001)]. Telomerase expression can actually make cells more resistant to apoptosis induced by oxidative stress [FEBS LETTERS; Ren,J; 488:133-138 (2001)].
Mice show no reduction of somatic cell telomere length with age [NATURE 347:400-402 (1990)] thanks to active somatic telomerase [SCIENCE 291:872-875 (2001)]. Telomeres in mouse stem cells do, however, shorten with age, possibly leading to decreased regenerative capacity [GENES & DEVELOPMENT; Flores,I; 22(5):654-667 (2008)]. Despite the apparent absence of somatic cell telomere shortening, most mouse somatic cells stop dividing after only 10−15 doublings. Possibly, in spite of the ultra-long telomeres on most chromosomes, a single chromosome with a short telomere could induce senescence [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Mark,J; 94(14):7423-7428 (1997)]. But mouse cells can become senescent despite being telomerase-positive. Nonetheless, transgenic mice with constitutively expressed TERT and enhanced expression of tumor-suppressor genes (to provide cancer resistance) have shown extended mean lifespan and reduced signs of aging [CELL; Tomas-Loba,A; 135(4):609-622 (2008)]. A study of 15 rodent species showed an inverse relationship between telomerase activity and body mass, but no relationship between telomerase activity and lifespan [AGE; Gorbunova,V; 30(2-3):111-119 (2008)].
Telomerase knockout mice (mice for whom the telomerase gene has been removed or "knocked-out") can sometimes maintain telomere length by a mechanism known as Alternative Lengthening of Telomeres (ALT). ALT can also occur in a human cell, but it is ten million times more likely to occur in a mouse cell [NATURE MEDICINE 6(8):849-851 (2000)]. ALT may be induced by p53 perturbations at telomeres [MOLECULAR AND CELLULAR BIOLOGY; Zaineb,R; 24(13):5967-5977 (2004)] or related to DNA repair at the site of the telomere.
PML bodies are donut-shaped protein aggregates in the nucleus containing PML (ProMyelocyclic Leukemia) protein along with other proteins such as pRb. PML bodies are suspected to normally play a role in tumor suppression. But in immortalized telomerase-negative ALT cells an aberrant form of PML bodies occur which contain telomeric DNA, telomere-binding proteins TRF1 & TRF2 and the Rad51 & Rad52 proteins that are normally active in homologous recombination repair of double-stranded DNA breaks [CANCER RESEARCH; Yeager,TR; 59(17):4175-4179 (1999)]. Extrachromosomal telomeric repeats found in the PML bodies can serve as templates for homologous recombination of telomeres [CANCER RESEARCH; Yeager,TR; 59(17):4175-4179 (1999)].
If cells continue to divide after having lost their telomeres (ie, beyond the Hayflick Limit of about 50 cell divisions), they not only become malfunctional due to lost genes, but the chromosome ends start sticking to other chromosomes — increasing the number of abnormalities. Typically a cell will invoke apoptosis ("cell suicide") or other or become senescent (stop the cell cycle) to prevent the cell from dividing or becoming cancerous. The Hayflick Limit itself may be a means of preventing cancer [SCIENCE; Campisi,J; 309:886-887 (2005)].
For those who believe that telomeres are a biological clock that cause aging by shortening, there has been the hope that human aging can be stopped by somehow adding active telomerase to all somatic cells. An experiment transfected human somatic cells with a reverse transcriptase subunit of telomerase thereby forcing the cells to express telomerase. The cells exhibited 20 population doublings beyond their Hayflick Limit and continued to exhibit normal, healthy and youthful cellular appearance & activity. This experiment was done not only for fibroblasts, but for retinal epithelial cells and vascular endothelial cells [SCIENCE 279:334&349 (1998)]. This result creates hope that it may someday be possible to preserve youth in some tissues by a form of gene therapy that either induces the expression of telomerase in somatic cells or adds additional genetic material to cells consisting of an engineered telomerase superior to the natural form. A person undergoing such therapy might first take a dose of telomerase destroyers to prevent any incipient cancers from being nourished by the treatment that would follow.
Nonetheless, only a few tissues that rapidly proliferate (endothelial cells, immune system cells, etc.) show decreased function with age that could be associated with telomere shortening. It is no accident that the notable exceptions to the rule of lack of telomerase in normal somatic cells are immune system cells [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Broccoli,D; 92(20):9082-9086 (1995)] and endothelial cells [CIRCULATION RESEARCH; Vasa,M; 87(7):540-542 (2000)]. For endothelial cells, the exhaustion of replicative capacity is greatest in areas of atherosclerosis — where the rate of cell division has been accelerated [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA)92(24):11190-11194 (1995)]. Telomere erosion contributes to defective liver regeneration and accelerated cirrhosis in chronic liver injury [SCIENCE; Rudolph,KL; 287:1253-1258 (2000)].
In one study, for a sample of 143 normal people over age 60 having shorter telomeres the chance of death was more than 3 times greater than average for heart disease and more than 8 times graater for infectious diseases, but there was no increased risk for cancer [THE LANCET; Cawthon,RM; 361:393-395 (2003)]. For males, telomere erosion has been correlated with unhealthy, pro-aging habits such as smoking, waist circumference, low physical activity and low fruit/vegetable intake [AGING CELL; Bekaert,S; 6(5):639-647 (2007) and AGING CELL; Huda,N; 6(5):709-713 (2007)]. Rate of telomere shortening has predicted cardiovascular mortality in the elderly [AGING; Epel,ES; 1(1):81-88 (2008)]. A study of 175 elderly Swedish twin-pairs found that the twins with the shortest telomeres (75% of the cohort) had 3 times the risk of death compared to the 25% with the longest telomeres [AGING CELL; Bakaysa,SL; 6(6):769-774 (2007)]. Other studies show no relationship between telomere length and morbidity or mortality in the elderly [AGING CELL; Martin-Ruiz,CM; 4(6):287-290 (2005) and EPIDEMIOLOGY; Bischoff,C; 17(2):190-194 (2006)].
People don't age or die because their cells aren't dividing. Cells in culture do not die after they cease dividing — and may survive as well as cells that never divide, such as neurons & muscle cells.
For background on cell cycle function, read Cell Cycle Control and Signalling Molecules and Transcriptions Factors.
The relationship between cellular aging and the aging of the whole organism is complex. Cellular "immortality" is essential for stem cells, but an "immortal" somatic cell is cancerous. Apoptosis (sometimes pronounced ap-ah-TOE-sis — the second "p" is silent) is programmed cell suicide — a genetically controlled cell death that causes cells to shrink and be eliminated without the tissue traumas associated with inflammation that accompanies uncontrolled cell death (necrosis). Whether a cell dies by apoptosis or necrosis is critically dependent upon the presence or absence of ATP [JOURNAL OF EXPERIMENTAL MEDICINE; 185(8):1481-1486 (1997)]. Apoptosis can benefit the organism by eliminating defective cells and protecting from cancer — or be associated with harmful conditions, as in atherosclerosis and neurogenerative disease. Cellular senescence (permanent cell cycle arrest) can benefit the organism by reducing vulnerability to cancer, but may also contribute to aging-associated tissue deterioration.
|
In apoptosis proteolytic enzymes (notably caspases — Cysteine ASpartase ProteASES) begin the process of orderly protein degradation that culminates in the production of small packages of cellular remnant. Apoptosis initiated by an extracellular signal (Fas receptor) activates caspase 8, whereas apoptosis due to intracellular damage or distress activates caspase 9. Both caspase 8 and caspase 9 are initiator caspases which can activate caspase 3, the primary effector caspase which induces apoptosis [JOURNAL OF NEUROCHEMISTRY; Polster,BM; 90(6):1281-1289 (2004)].
The tumor-suppressor protein p53 can be a potent initiator of apoptosis, whereas anti-apoptotic Bcl−2 is an oncogene because mutations in the gene increase Bcl−2 protein expression, thereby protecting cancer cells from apoptosis. There is a "family" of Bcl−2 proteins, all of which possess at least one of four Bcl−2 Homology domains (BH1 to BH4). The anti-apoptosis subfamily (which includes Bcl−2, Mcl−1 and Bcl−xL) have all of the homology domains, whereas the pro-apoptotic subfamily (Bax, Bak, Bad, Bim, Bid, Bik, PUMA, Noxa, etc.) are all missing BH1. Bim, Bad, Bid, PUMA and Noxa only contain BH3 [SCIENCE; Adams,JM; 281:1322-1324 (1998)]. In response to DNA damage, PUMA (p53 Upregulated Mediator of Apoptosis) mediates Bax translocation to the mitochondria [JOURNAL OF BIOLOGICAL CHEMISTRY; Melino,G; 279(9):8076-8083 (2004)]. In response to DNA damage p53 protein can induce apoptosis by increasing transcription of BH3-only proteins [SCIENCE; Villuger,A; 302:1036-1038 (2003)]. Anti-apoptotic members of the Bcl−2 family stabilize the outer mitochondrial membrane (preventing cytochrome−c release) whereas pro-apoptotic members increase permeability of the outer mitochondrial membrane [BMC CELL BIOLOGY; Lutter,M; 2:22-30 (2001)]. Bax and Bak are restrained from permeabilizing the mitochondrial membrane by their association with the anti-apoptotic subfamily proteins. The BH3-only proteins promote apoptosis by interfering with the association of the anti-apoptotic subfamily proteins with Bax and Bak [SCIENCE; Willis,SN; 315:856-859 (2007)], and (in the case of Bid & Bim) by directly activating Bax & Bak) [NATURE; Gavathiotis,E; 455:1076-1081 (2008)].
If intracelluar Ca2+ is high, p53 may be bypassed because high mitochondrial
Ca2+ opens the
Mitochondrial Permeability Transition Pore (MPTP)
causing energy uncoupling (reduced inner membrane proton gradient), increased superoxide
production, reduced ATP production and the release of cytochrome c to the
cytosol — which activates caspase 9. Caspase 9 activates caspase 3 and
caspase 7 by forming an apoptosome with cytochrome−c and Apoptotic
Protease Activating Factor−1 (APAF−1). Oxidative stress, DNA damage
and cell stress other than high Ca2+ may induce Bid protein to
form Bax/Bak channels and release of
cytochrome−c [JOURNAL OF BIOLOGICAL CHEMISTRY; Rostovtseva,TK; 279(14):13575-13583
(2004)].
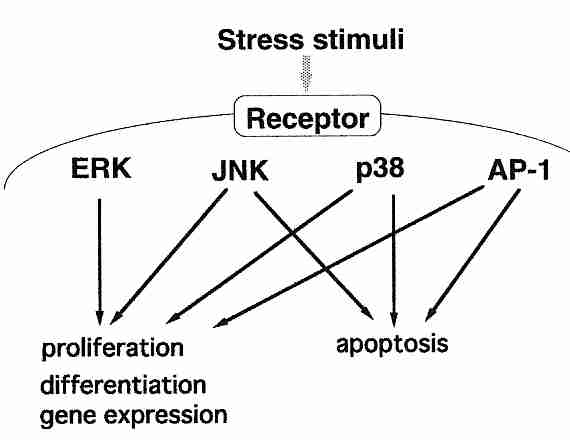
|
Mitogens are agents that trigger mitosis (cell division). Growth factors are mitogens, but stress can be mitogenic as well. Although very active cell proliferation (mitosis) is essential to growth & development in a young organism, in an older organism proliferation may often be associated with inflammation. Proliferation in older animals more easily leads to cancer (short-lived invertebrates usually have post-mitotic cells). Mitogens generally act at cell surfaces, and cell signalling resulting from surface stimulation is by Mitogen Activated Protein Kinases (MAPKs). (A kinase is an enzyme that transfers a phosphate group from ATP, GTP, ADP, etc, to an enzyme, thereby activating the enzyme. A phospatase does the opposite, inactivating enzymes by removing a phosphate group.) MAPK pathways are typically a series of kinases that activate other kinases.
There are three families of MAPKs: (1) Extracellular signal-Regulated Kinases (ERKs), (2) c−Jun N−terminal Kinases (JNKs) and (3) the p38 family of kinases. The ERK family responds to growth factors, resulting in proliferation & differentiation, whereas the other two families respond to a variety of stresses or inflammatory cytokines that can lead either to apoptosis or to proliferation — depending on the tissue & stimulation. The most important inflammatory kinase is p38. Activator Protein−1 (AP−1, a regulator of cell survival and proliferation) is a transcription factor activated by either ERK or JNK. AP−1 can be pro-apoptotic or anti-apoptotic, but is most often anti-apoptotic (in association with DNA-repair). ATM loss leads to JNK-mediated AP−1 stress [JOURNAL OF BIOLOGICAL CHEMISTRY; Weizman,N; 278(9):6741-6747 (2003)].
Senescent cells (cells that no longer proliferate or divide in response to growth factors or mitogens) can function like normal cells, but display a number of distinctive characteristics. Some of these characteristics, such as increased free radical production, increased oxidative damage, increased glycation damage and reduced heat shock protein expression may simply be due to the fact that senescent cells are usually "old". Senescent cells completely lack H1 histone (the histone which causes second-order compacting of chromatin) and contain transcriptionally silent heterchromatic foci which are believed to repress proliferation genes [JOURNAL OF CELL BIOLOGY; Funayama,R; 175(6):869-880 (2006)].
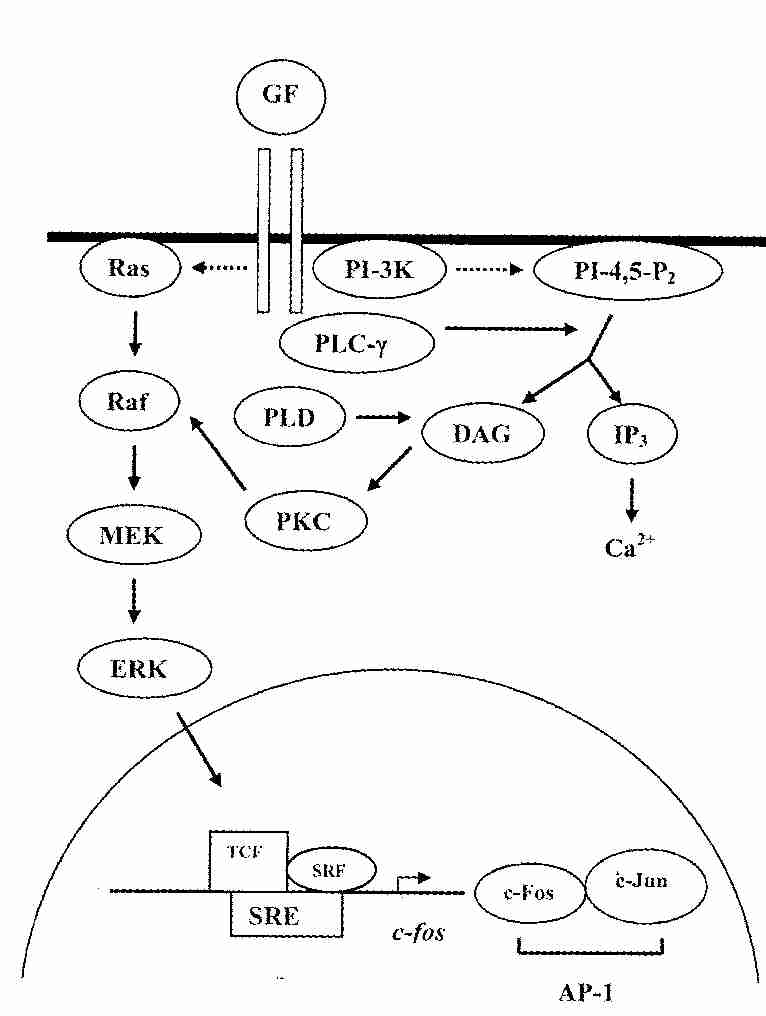
|
But the accumulation of defective proteins may be partially due to a genetic down-regulation of proteasome activity associated with the senescent phenotype [JOURNAL OF BIOLOGICAL CHEMISTRY; Chondrogianni, N; 278(30):28026-28037 (2003)]. Although Growth Factors (GFs) can still activate the Ras/Raf/MEK/ERK pathway in senescent cells, the ERK subgroup of MAPKs fail to enter the nucleus, c−fos induction is reduced and Activator Protein−1 (AP−1) transcription factor is far less capable of binding to DNA. AP−1 activity as a regulator of cell survival — proliferation is highly influenced by the AP−1 consitituents [JOURNAL OF CELL SCIENCE; Hess,J; 117(Pt 25); 5965-5973 (2004)]. Depending on the influence of other transcription factors, c−fos can cause cellular proliferation, differentiation or apoptosis. Serum Response Elements (SREs) regulate c−fos expression, which is activated by the Ternary Complex Factor (TCF) transcription factors that cannot bind to SRE without Serum Response Factor (SRF). The c−Jun protein is activated by the Jun Kinase (JNK) MAPKs. JNK activity is most stimulated by UV light, whereas ERKs are most strongly stimulated by growth factors [JOURNAL OF BIOLOGICAL CHEMISTRY; Karin,M; 270(28):16483-16486 (1995)]. Increased expression of c−Jun due to ultraviolet light leads to AP−1 induction of metalloproteinases (collagenases) that contribute to "photoaging" of skin [JOURNAL OF CLINICAL INVESTIGATION; Fisher,GJ; 101(6):1432-1440 (1998)].
Senescent cells are resistant to apoptosis, unlike the postmitotic neurons that apoptotically contribute to neurodegeneration. Senescent cells are not only more sensitive to cell injury, they have larger nuclei and less regular shape. Senescent fibroblasts secrete metalloproteinases that degrade the collagen matrix secreted by normal fibroblasts. Senescent fibroblasts also secrete inflammatory cytokines, such as InterLeukin−1 (IL−1).
Resistance of aging cells to apoptosis may be due to a decline in apoptotic protein function rather than cell senescence [NATURE MEDICINE; Suh,Y; 8(1):3-4 (2002)]. Caloric Restriction with Adequate Nutrition (CRAN) increases rat liver cell apoptosis, particularly for pre-cancerous cells [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA) 91(21):9995-9999 (1994)]. Normally reduced proteolysis by proteasomes allows p53 accumulation to induce apoptosis [JOURNAL OF BIOLOGICAL CHEMISTRY; Lopes,UG; 272(20):12893-12896 (1997)], but the mechanisms become more dysfunctional with aging. In senescent cells proteasome activity declines even more rapidly, resulting in a faster accumulation of undegraded protein products [THE FASEB JOURNAL; Sitte,N; 14(15):2495-2502 (2000)].
One could easily imagine that the accumulation of increasing numbers of senescent cells within tissues would contribute to aging of tissues & organs. Although this appears to be the case in diabetics and progeria victims (Werner's Syndrome, Down's Syndrome and childhood progeria), it has not been demonstrated for fibroblasts of "healthy" persons [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Cristofalo,VJ; 95(18):10614-10619 (1998)]. Only a minority of fibroblasts are senescent in the healthy elderly. On the other hand, cellular senescence may play a critical role in aging endothelial cells and the development of atherosclerosis [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Chang,E; 92(24):11190-11194 (1995)] and in T−cells.
In mammals, shortening of telomeres leads to senescence in some cells (e.g., fibroblasts) and apoptosis in other cells (e.g., T−cells) [SCIENCE; Karlseder,J; 283:1321-1325 (1999)]. The ATM protein kinase which activates p53 protein in response to DNA damage also activates p53 in response to telomere shortening. Inhibition of ATM in DNA damage conditions leads to reduced cell senescence and increased apoptosis [JOURNAL OF BIOLOGICAL CHEMISTRY; Zhang,X; 280(20):19635-19640 (2005)]. Other proteins which participate in NHEJ of double-strand break (DSB) repair assist in apoptosis induction if DSB repair fails [CELL SIGNALLING; Abe,T; 20(11):1978-1985 (2008)]. The cell cycle is halted by p21Cip1 protein (activated by p53), which initiates cell senescence. But p21 is only expressed transiently. Long-term maintenance of cellular senescence requires expression of the cell cycle inhibitor p16INK4a protein, which is also induced by p53.
A study of rodent organs found an average 10-fold increase in p16INK4a expression and an average of 3.5-fold increase in Arf expression with age, concluding that these proteins are biomarkers — and possible effectors — of both cellular senescence and of mammalian aging [JOURNAL OF CLINICAL INVESTIGATION; Krishnamurthy,J; 114(9):1299-1307 (2004)]. Increased p16INK4a expression with age may lead to increased senescence of pancreatic β−cell stem cells in non-insulin-resistant type 2 diabetes [NATURE; Krishnamarthy,J; 443:453-457 (2006)] — and increased stem cell senescence associated with declining neurogenesis in some (but not all) areas of the brain [NATURE; Molofsky,AV; 443:448-452 (2006)]. In human T-lymphocytes, p16INK4a expression increases with age between ages 20 to 80, with twice the increase in smokers [SCIENCE; Liu,Y; 8(4):439-448 (2009)]. Removal of p16INK4a-positive senescent cells in mice delayed the onset of age-related pathologies [NATURE; Baker,DJ; 479:232-236 (2011)]. Mice that were transgenic with extra genes of both p53 and Arf (with normal activity of both) had strong cancer resistance, increased levels of reduced glutathione (GSH) antioxidant, and lifespan increase of 16% [NATURE; Matheu,A; 448:375-380 (2007)].
Both p16 and p21 act by reducing pRB phosphorylation, thereby preventing expression of the EF2 transcription factors required for DNA synthesis. Nonetheless, the p53 and pRb tumor-suppressor proteins make partially independent contributions to cellular senescence. Exposure of the telomere 3' overhang after telomere loop disruption appears to be the critical signal for replicative senescence because oligonucleotides with this overhang can induce senescence in fibroblasts [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Li,G; 100(2):527-531 (2003)].
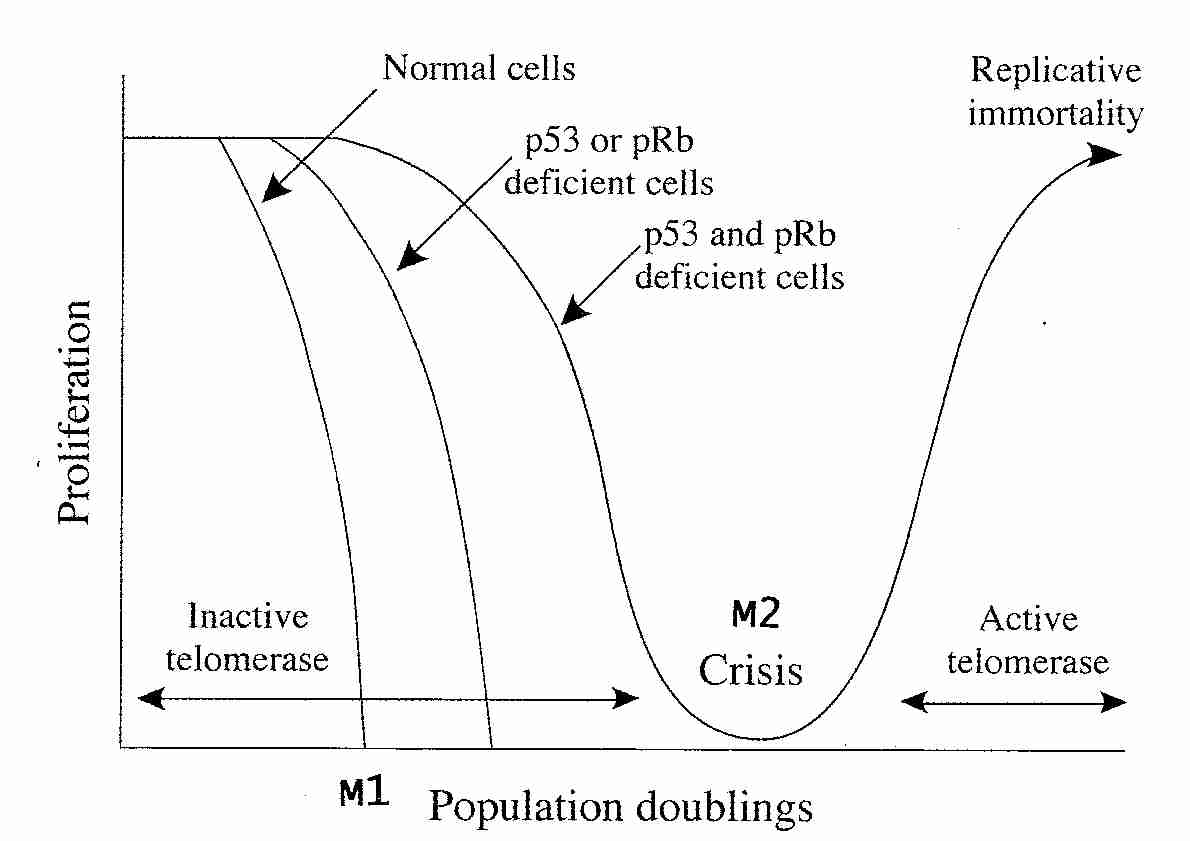
|
In normal human cells, telomere shortening (typically 50−200 base pairs lost per cell division) will induce cellular senescence after between 50 to 100 cell divisions (in vitro population doublings), depending on the cell type. Normal cellular senescence is designated M1 (Mortality Stage 1). If either p53 or pRb expression is inhibited (eg, through defective genes), senescence will occur after about ten additional population doublings. If both p53 and pRb expression is inhibited (eg, by simian virus 40, SV40), then about twenty additional doublings will occur and cells enter M2 (Mortality Stage 2), also called crisis. In contrast to M1 cells (which have short telomeres of about 4 Kbp, Kilobase pairs), M2 cells have extremely short telomeres (about 1.5 Kbp), are genetically unstable and usually die quickly [THE EMBO JOURNAL; Counter,CM; 11(5):1921-1929 (1992)]. Fewer than one in a million cells survive crisis. Surviving cells nearly always become "immortalized" by telomere expression.
(In contrast to humans, although rats display senescence for fibroblasts, they show no sign of replicative senescence for glial cells [SCIENCE; Mathon,NF; 291:872-875 (2001)].)
So-called premature cellular senescence can be provoked by various sublethal cellular stresses such as hydrogen peroxide, ultraviolet irradiation and similarly damaging agents which either accelerate the number of telomeres lost per division or directly induce DNA damage or both. Fenton reaction-mediated DNA damage is seven times more likely to occur in telomeres than elsewhere in a chromosome, probably because of the higher proportion of guanosines in the telomere (TTAGGG) [JOURNAL OF BIOLOGICAL CHEMISTRY; Henle,ES; 274(2):962-971 (1999)]. In fact, the role of oxidation in telomere shortening is dramatically demonstrated by the fact that spin-trapping agent PBN treatment of cells can increase their number of population doublings by as much as 25% [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Chen,Q; 92(10):4337-4341 (1995)].
But premature cellular senescence can also be induced by agents that are more directly involved in cell signalling dysfunction, such as ras oncogene overexpression [CELL; Serrano,M;88(5):593-602 (1997)], PML protein overexpression [THE EMBO JOURNAL; Bischof,O; 21(13):3358-3369 (2002)], Transforming Growth Factor beta (TGF−ß) or histone deacetylase inhibition [MOLECULAR AND CELLULAR BIOLOGY; Ogryzko,VV; 16(9):5210-5218 (1996)]. Stress-induced senescence due to Ras protein requires signalling from the p38 (stress-activated) form of Mitogen-Activated Protein Kinase (MAPK) and cannot be prevented by hTERT-mediated telomerase elongation. TGF−ß inhibits telomerase and, like Ras protein, mediates senescence by p38 MAPK activation. Cellular senscence due to DNA damage can be triggered by p38 MAPK signalling and does not require ATM protein [JOURNAL OF BIOLOGICAL CHEMISTRY; Naka,K; 279(3):2030-2037 (2004)].
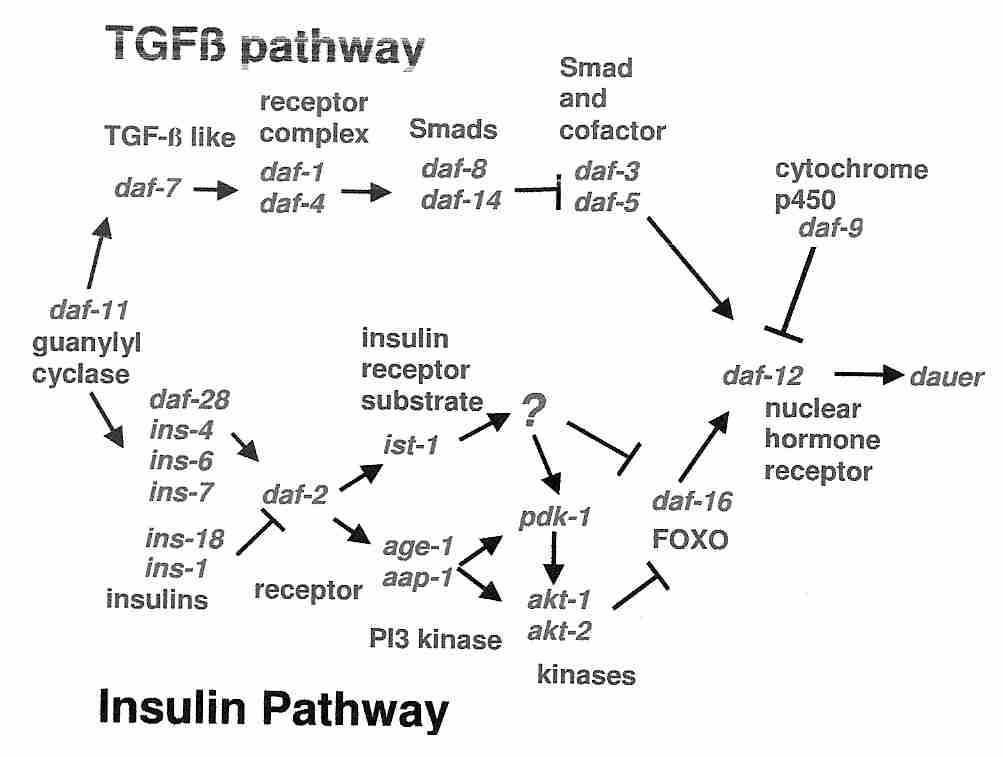
|
Homeostasis and remodeling of the extracellular matrix is mediated in large part through an interplay of Matrix MetalloProteinases (MMPs, collagenase) and Tissue Inhibitors of MetalloProteinases (TIMPs). Senescence of fibroblasts is accompanied by a shift from matrix synthesizing to matrix degredation associated with increased MMP production and decline of tissue function. TGF−ß can induce either apoptosis [ENDOCRINOLOGY; Bruckheimer,EM; 142(6):2419-2426 (2001)] or senescence [ENDOCRINE-RELATED CANCER; Fleisch,MC; 13(2):379-400 (2006)]. Peculiarly, TGF−ß induces TIMP and represses MMP [JOURNAL OF BIOLOGICAL CHEMISTRY; Hall,M; 278(12):10304-10312 (2003)]. Also peculiar is the fact that MMP production seems to be mediated by the same forkhead family of transcription factors that are credited with increased longevity in C. elegans nematode worms due to disruption of insulin/IGF1−1-like signalling [JOURNAL OF BIOLOGICAL CHEMISTRY; Mawal-Dewan; 277(10):7857-7864 (2002)] and that mutations in the TGF−ß pathway can induce C. elegans dauer formation, but (unlike mutations in the insulin/IGF−1 pathway) do not extend adult nematode lifespan [BMC DEVELOPMENTAL BIOLOGY; Liu,T; 4:11 (2004)]. (See LONGEVITY GENES (FLIES & WORMS) for more about C. elegans dauer formation and lifespan extension associated with insulin/IGF−1 signalling.)
Both p53 and pRB participate in apoptosis as well as senescence. Whereas p53 induces apoptosis in response to DNA damage, loss of pRB leads to apoptosis and deregulated cell proliferation [CURRENT OPINION IN GENETICS & DEVELOPMENT; Hickman,ES; 12(1):60-66 (2002)]. The p53 protein not only induces apoptosis by increasing gene expression of Bax, Bak and a number of other proteins [BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS; Michalak,E; 331(3):786-798 (2005)], but p53 also directly activates Bax protein in the cytoplasm [SCIENCE; Chipuk,JE; 303:1010-1014 (2004)].
What is the relationship between cellular senescence, apoptosis, cancer and aging? Apoptosis in development is the reason humans do not have webbed hands. In the developing nervous system cell proliferation accompanies apoptosis with "survival of the fittest" synaptic connections. The great majority of T−lymphocytes produced are eliminated by apoptosis, an important defense against auto-immune disease. T−cells express a Fas (CD95) receptor which mediates an apoptotic signal that bypasses nuclear transcription and directly activates proteases, thus terminating the immune response. Fas receptor expression on T−cells increases with aging, enhancing the susceptibility of T−cells to apoptosis [CELL AND TISSUE RESEARCH; Higami,Y; 301(1):125-132 (2000)].
A youthful, healty organism has efficient cell-cycle control and can thereby resist undesirable apoptosis while efficiently using apoptosis when needed. Cells having DNA defects or mitochondria producing excessive free radicals can be eliminated by apoptosis and macrophages without causing inflammation. Aged cells with less effective cell-cycle control will less readily apoptose when defective, but will more often dysfunctionally apoptose. High levels of apoptosis in aged tissues result in tissue degeneration. Accumulated free radical, glycation and other forms of cellular damage lead increasingly to dysfuncional cell-cycle control with age. Some of those immersed in genetic paradigms of aging assert that CRAN (Caloric Restriction with Adequate Nutrition) "up-regulates" apoptosis in cancer cells while "down-regulating" apoptosis in normal cells. A more reasonable explanation might be that by reducing oxidative stress & glycation, CRAN maintains youthful cell-cycle control.
Apoptosis may be protective in some tissues, whereas cellular senescence may be more protective against cancer in other tissues. Reduction of nitric oxide synthesis with aging reduces the nitric oxide inhibition of endothelial cells apoptosis — leading to a worsening of atherosclerotic disease. Endothelial cells have a high rate of telomere loss and senescent endothelial cells contribute to atherosclerosis by the release of pro-inflammatory cytokines [CIRCULATION; Minamino,T; 105(13):1541-1544 (2002)].
The "longevity gene" SIRT1 gene silencing protein increases cell cycle arrest by FOXO transcription factor while inhibiting FOXO's induction of apoptosis [SCIENCE; Brunet,A; 303:2011-2015 (2004)]. The p53 protein arrests cell growth (cell cycle arrest) & triggers cell suicide (apoptosis) — typically as a response to DNA damage. Normally p53 protein induction of cellular senescence (halted growth cycle) is regarded as a defense against cancer, but SIRT1 inhibition of p53-mediated apoptosis and cell senescence is presumed to be life-extending by allowing for cell repair.
Cellular senescence has been called an "antagonistic pleiotropic trait" that benefits young organisms at the expense of harm to older organisms. An exaggerated example of this may be p53+/m mutant mice, which show enhanced p53 protein activity. Although the mutant mice show an accelerated aging phenotype and only live 80% as long as normal mice, cancer is exceedingly rare in these mutants [NATURE; Tyner,SD; 415:45-53 (2002)]. The mice support the views that cellular senescence/apoptosis is a defense against cancer and that cellular senescence/apoptosis can lead to senescence (aging) of the organism as a whole. Cellular senescence may also be antagonistically pleiotropic due to the secretions of senescent cells, which have been shown to promote cancer growth in surrounding tissues [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Krtolica,A; 98(21):12072-12077 (2001)].
Werner's Syndrome (reputedly the segmental progeria most resembling accelerated aging) is often characterized as a disease of accelerated telomerase functioning and cellular senescence. In contrast to normal human fibroblasts which senesce after about 50 population doublings, Werner's patients' fibroblasts usually senesce after about 20 doublings — with longer than normal telomeres. The WRN protein, which is defective in Werner's patients is a helicase (enzyme that unwinds double helical regions in DNA and RNA) and an exonuclease (enzyme that catalyzes hydrolytic removal of nucleotides from the end of DNA and RNA).
Werner's patient fibroblasts are resistant to apoptosis. The high proportion of sarcomas in those patients may be due to the promotion of transformation of the non-senescent cells by the proteases & cytokines created by the many senescent cells. Werner's Syndrome is not simply a model system of the effect of a high proportion of senescent cells because defective DNA repair plays such a prominent role in the disease.
Transgenic mice that have defective repair of mitochondrial DNA will have reduced lifespan, increased apoptosis and display an "accelerated aging" phenotype [SCIENCE; Kujoth,GC; 309(5733):481-484 (2005)]. The mice show no sign of oxidative stress or increased free radical production.
In general, apoptosis can be described as being signal-induced or damage-induced. Signal-induced apoptosis is essential for the precision elimination of undesirable cells following proliferation of many cell types, including clonal expansion of T−cells. Well-controlled apoptosis is a feature of an efficient immune system. Damage-induced apoptosis is a major factor in neurodegeneration, although the process undoubtedly becomes increasingly necrotic. The same may be true for macrophages that die in atherosclerotic plaques. If the immune system induces apoptosis in cancer cells, it could involve both forms of apoptosis. There is a decrease in both kinds of apoptosis with age, as cellular signalling and regulation (including apoptotic regulation) becomes less efficient. A tissue deprived of many cells because of a high level of apoptosis may display the "aged phenotype" as much as a tissue composed largely of senescent cells that are too defective to undergo apoptosis.
A biomarker of cell senescence would facilitate the identification and study of senescent cells, as well as the targeting for destruction of such cells. The most promising candidate biomarker, ß-galactosidase, is elevated in replicative senescence and can quantitatively estimate replicative age in vitro. But cellular ß-galactosidase also is present in immortal cells and can be induced by subjecting cells to hydrogen peroxide [EXPERIMENTAL CELL RESEARCH; Severino,J; 257:162-171 (2000)].
It has been suggested that reprogramming cells to apoptose rather than senesce may be a means to reduce cancer and eliminate one cause of aging.
"Accelerated aging" is distinct from accelerated mortality because "accelerated aging" diseases exhibit an elderly phenotype and increased disposition to aging-associated diseases such as cancer and Alzheimer's Disease. Too often, however, "accelerated aging" is equated with increased disposition to aging-associated diseases in the absence of an elderly phenotype). High blood pressure and AIDS accelerate mortality without exhibiting an elderly phenotype. Without objective biomarkers of aging the "elderly phenotype" is open to dispute.
No disease condition displays all symptoms of accelerated aging. Diseases that resemble certain aspects of accelerated aging are known as segmental progerias, because of the "segments" of aging in each disease condition. If aging is due to a variety of cellular and molecular damages, segmental progerias may represent subsets of those damages.
Segmental progerias primarily are diseases of defective DNA-repair, although diabetics also show many features of accelerated aging. Defects in Base Excision Repair (BER), however, are generally too lethal to manifest as accelerated aging [SCIENCE; Hasty,P; 299:1355-1359 (2003)]. (Genomic instability syndromes are not necessarily progerias.)
It has been proposed that segmental progerias result from decreased cytotoxic DNA damage repair or from an exaggerated response to DNA damage signals — whereas cancers result from decreased mutagenic DNA damage repair or from an impaired response to DNA damage signals — with excision repair mainly effective against cancer and transcription-coupled (or interstrand cross-link) repair mainly facilitating longevity [CURRENT OPINION IN CELL BIOLOGY; Mitchell,JR; 15(2):232-240 (2003)]. Progeroid syndromes have been associated with NER and DSB repair, but not with BER or MMR [JOURNAL OF INTERNAL MEDICINE; Lombard,DB; 263(2):128-141 (2008)]. In both human patients and mouse models only some DNA repair defects show accelerated aging, namely the TCR subtype of NER and defects to NHEJ genes. The GGR subtype of NER mainly results in increased carcinogenesis and "photo-aging", although there is neurodegeneration. BER defects are generally lethal [SCIENCE; Hasty,P; 299:1355-1359 (2003)], but SIRT6 (which facilitates BER by an unknown mechanism) knockout mice do show an accelerated aging phenotype (including loss of subcutaneous fat and decreased bone density) during the few weeks in which they are able to survive [Mostoslavsky, 2006].
Werner's syndrome (WS) is associated with early onset of very many age-related diseases and most closely represents accelerated aging of any of the segmental progerias. About two-thirds of WS victims are Japanese (attributed to inbreeding). WS is due to a defect or deletion of a single gene (WRN), resulting in defects of both telomeres and DNA repair [MOLECULAR AND CELLULAR BIOLOGY; Du,X; 24(19):8437-8446 (2004)]. The WRN gene is a member of the helicase family that causes DNA to unwind, which is a requirement for most forms of DNA repair. Defective WRN protein results in a reduction of p53-mediated apoptosis. There is an accelerated rate of somatic mutations, particularly deletions [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Fukuchi,K; 86(15):5893-5897 (1989)], although defects are also probable in BER and NHEJ [MECHANISMS OF AGEING AND DEVELOPMENT; Kusumoto,R; 128(1):50-57 (2007)]. Defective homologous recombination is believed to be the primary reason for the chromosomal abnormalities of WS victims [GENES & DEVELOPMENT; Prince,PR; 15(8):933-938 (2001) and MOLECULAR AND CELLULAR BIOLOGY; Saintigny,Y; 22(20):6971-6978 (2002)]. Defective recombination leads to genomic instability and thus greatly increased risk of cancer, particularly sarcomas (the relative incidence of mesenchymal cell cancer compared to epithelial cell cancer is ten times normal) [CANCER EPIDEMIOLOGY, BIOMARKERS & PREVENTION; Goto,M; 5(4):239-246 (1996)]. WRN has been described as a tumor-suppressor gene because epigenetic silencing of WRN increases chromosomal instability and because tumor-types match those of other tumor-suppressor gene defects [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Agrelo,R; 103(23):8822-8827 (2006)]. But WRN also acts as a tumor suppressor by its facilitation of p53-mediated apoptosis [GENES & DEVELOPMENT; Spillare,EA; 13(11):1355-1360 (1999)]. Unlike carcinomas, mesenchymal tumors primarily maintain telomeres by the ALT mechanism [JOURNAL OF CELL SCIENCE; Multani,AS; 120(Pt 5):713-721 (2007)]. Although mesenchymal malignancies predominate for both mice & WS victims, WRN knockout mice show no sign of accelerated aging despite reduced cellular proliferation capacity [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Lebel,N; 95(22):13097 (1998)]. Whereas normal human fibroblasts experience replicative senescence after about 60 divisions, the fibroblasts of WS patients senesce after about 20 divisions [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Faragher,RGA; 90(24):12030-12034 (1993)]. The accelerated senescence of fibroblasts from WS patients is associated with an accelerated accumulation of double-strand breaks [JOURNAL OF RADIATION RESEARCH; Ariyoshi,K; 48(3):219-231 (2007)]. Telomeres are of normal length initially, but shorten at an abnormally high rate, resulting in many so-called senescent cells (creating a model system for the study of the senescent cell phenotype). Telomere repair is reduced, as is MisMatch Repair (MMR), TCR Base Excision Repair (BER) and double-strand break repair of DNA. Transcription of mRNA by RNA polymerase II from DNA is roughly half as efficient in WS cells compared to normal cells [MOLECULAR BIOLOGY OF THE CELL; Balajee,AS; 10(8):2655-2668 (1999)]. The disease first becomes evident in the late teens or early 20s, and typically results in death by age 50 by cardiovascular disease. Osteoporosis, premature hair graying, alopecia, high blood pressure, stroke, cataracts, severe atherosclerosis, and type 2 diabetes are extremely common [MECHANISMS OF AGING & DEVELOPMENT; Goto,M; 98(3):239-254 (1997)]. Many of these effects may be due to increased levels of the inflammatory cytokines produced by aging and senescent cells [EXPERIMENTAL GERONTOLOGY; Kumar,S; 28(6):505-513 (1993) and REJUVENATION RESEARCH; Davis,T; 9(3):402-407 (2006)]. Conversely, proinflammatory cytokines have been shown to induce cellular senescence [FREE RADICAL RESEARCH; Sasaki,M; 42(7):625-632 (2008)]. Abnormally high levels of collagenase from senescent fibroblasts leads to loss of skin elasticity and to skin wrinkling. Experimentally produced cellular senescence in rat arteries results in an atherosclerotic phenotype matching that seen in WS and normal aging [CIRCULATION; Minamino,T; 108(18):2264-2269 (2003)], which supports the general contention that cell senescence contributes significantly to the normal aging phenotype. At a much faster rate than is often seen in normal aging, WS victims accumulate visceral fat, develop high levels of the plasma cytokine TNF−α, develop insulin resistance and show a metabolic syndrome profile, but without being obese [DIABETES CARE; Yokote,K; 27(10):2562-2563 (2004)]. The carbonyl content of proteins in WS victims increases exponentially with age at a much higher rate than normal. Microarray profiling of 6,912 human fibroblast genes showed 91% of the genes were common to WS and aging cells, 6% were unique to normal aging and 3% were unique to WS [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Kyng,KJ; 100(21):12259-12264 (2003)]. But WS victims show no increased tendency for neurodegeneration, prostate problems or Alzheimer's Disease — and the immune system remains normal. The progeroid symptoms of WS have been attributed to both increased cellular senescence [SCIENCE; Kipling,D; 305:1426-1431 (2004)] and increased apoptosis [MOLECULAR BIOLOGY OF THE CELL; Pichierri,P; 12(8):2412-2421 (2001)].
In Hutchinson-Gilford Progeria Syndrome (HGPS, "childhood progeria", in contrast to the "adult progeria" of Werner's syndrome) a child is born with abnormally short telomeres. Childhood progeria occurs once per 4−8 million births. Victims are characterized by short stature, early hair loss, cardiovascular problems (stroke and coronary dysfunction are common) and an elderly facial phenotype, but normal cognition and immune function, and no disposition to cancer [AMERICAN JOURNAL OF MEDICAL GENETICS (PART A); Hennekam,RCM; 140(23):2603-2624 (2006)]. The disease is caused by a point mutation in the gene for lamin A, a filament protein in the nuclear matrix and nuclear lamina that is required for DNA replication and nuclear organization. The point mutation results in a prelamin A protein called progerin that cannot be converted to lamin A because it is missing 50 amino acids. Progerin retains a hydrophobic farnesyl group (normally cleaved by the protease ZMPSTE24) which causes it to be highly membrane-associated. The intranuclear scaffold formed by lamins may facilitate transcription, replication and DNA repair [NATURE MEDICINE; Liu,B; 11(7):780-785 (2005)]. Disruption of nuclear lamin organization inhibits mRNA transcription (RNA polymerase II activity) in mammalian cells [THE JOURNAL OF CELL BIOLOGY; Spann,TP; 156(4):603-608 (2002)]. DSBs accumulate in HGPS and ZMPSTE24-deficient cells, where DSB-repair is apparently blocked by accumulation of mis-localized XPA protein at the damage site [THE FASEB JOURNAL; Liu,Y; 22(2):603-611 (2008)]. Cells with the lamin A mutation show an impaired ability to form foci for the recruitment of DNA repair proteins during DNA replication, resulting in defective homologous recombination [NATURE MEDICINE; Liu,B; 11(7):780-785 (2005) and JOURNAL OF CELL SCIENCE; Manju,K; 119(Pt 13):2704-2714 (2006) and DNA REPAIR; Paulson;RD; 6(7):953-966 (2007)]. At age 5 the telomeres of a Hutchinson-Gilford syndrome child are about as long as those of a very elderly person. HGPS patient cells show loss of epigenetic control [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Shumaker,DK; 103(23):8703-8708 (2006)]. A study which compared HGPS patient cells with the skin cells from young & elderly human subjects found similar defects in the HGPS & elderly cells, including down-regulation of certain nuclear proteins, increased DNA damage and demethylation of H3 histone leading to reduced heterochromatin, suggesting that lamin A defects contribute to normal aging [SCIENCE; Scaffidi,P; 312:1059-1063 (2006)]. Nuclear levels of phosphorylated H2AX — which recruits DNA repair proteins to sites of DNA damage [SCIENCE; Celeste,A; 296:992-997 (2005)] — is more than three times higher in fibroblasts from old (81 to 96 years) as compared to young (3 to 11 years) normal humans [SCIENCE; Scaffidi,P; 312:1059-1063 (2006)], comparable to what is seen in HGPS cells [NATURE MEDICINE; Liu,B; 11(7):780-785 (2005)]. Most often these children die of myocardial infarction or stroke (average age of death is 13). The premature atherosclerosis is without the usual causes association with high blood pressure or high blood cholesterol. Progestin preferentially accumulates in the nuclei of vascular smooth muscle and endothelial cells causing the cells to senesce or become apoptotic [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); McClintock,D; 103(7):2154-2159 (2006)]. Carbonyl content of protein rises more rapidly with age than in any of the other segmental progerias. These children do not have the high rates of presbyopia, cataracts, osteoporosis or Alzheimer's Disease often seen in the elderly. Like WS, HGPS is primarily a disease of proliferative tissues characterized by high rates of cellular senescence and apoptosis [EXPERIMENTAL GERONTOLOGY; Bridger,JM; 39(5):717-724 (2004)].
Bloom's syndrome, like Werner's syndrome, is due to a defective helicase-type protein, in this case BLM — leading to chromosome aberrations [NUCLEIC ACIDS RESEARCH; Brosh,RM; 35(22):7527-7544 (2007)]. The disease is most common in Ashkenazi Jews (descendents of Eastern European Jews) due to intensive inbreeding. Victims are small at birth and rarely grow to be taller than 5 feet. Intelligence is usually normal. Photosensitivity causes the face to be red. Approximately 10% of victims have type 2 diabetes. Immunodeficiency leads to recurrent severe infections of the respiratory tract and ear. Women have reduced fertility and men are usually completely infertile. Death is most often due to cancer. If they survive death from leukemia at an average age of 22, Bloom's victims usually die of solid tumors at an average age of 35. BLM protein is preferentially expressed in proliferative tissues containing high levels of telomerase. BLM protein deficiency renders cells highly vulnerable to p53-induced apoptosis, which is suggested to contribute to growth retardation. During the S-phase of the cell cycle BLM protein localizes in the nucleolus where it apparently assists in the resolution of stalled replication forks [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Yankiwski,V; 97(10):5214-5219 (2000)]. The BLM protein is normally found mainly in the PML bodies of the nucleus, but when NHEJ is required to repair DSBs, BLM protein causes rapid recruitment of BRCA1 protein to the site of damage [JOURNAL OF CELL BIOLOGY; Davalos,AR; 162(7):1197-1209 (2003)]. The high rate of apoptosis in Bloom's Syndrome victims would be predictive of an accelerated aging phenotype with reduced cancer, but high proliferation & decreased genome stability selects for p53 mutations which reduce apoptosis & foster cancer [JOURNAL OF CELL BIOLOGY; Davalos,AR; 162(7):1197-1209 (2003)]. BLM protein highly co-localizes with telomeres in cells maintaining telomere length by the ALT mechanism, but not in normal or telomerase-positive cells [HUMAN MOLECULAR GENETICS; Stavropoulos,DJ; 11(25):3135-3144 (2002)]. Chromosomal breaks and a greatly elevated rate of sister chromatid exchanges are characteristic features of Bloom's syndrome [AMERICAN JOURNAL OF HUMAN GENETICS; German,J; 29(3):248-255 (1977)]. BLM protein prevents excessive homologous recombination [NATURE; Wu,L; 426:870-874 (2003)], particularly where D−loops are formed [NUCLEIC ACIDS RESEARCH; Bachrati,CZ; 34(8):2269-2279(2006)]. BLM protein also complexes with the Rad51 protein in homologous recombination repair of double-strand DNA breaks [JOURNAL OF BIOLOGICAL CHEMISTRY; Braybrooke,JP; 278(48):48357-48366 (2003)] and inhibits the exonuclease activity of WRN protein [JOURNAL OF BIOLOGICAL CHEMISTRY; von Kobbe,C; 277(24):22035-22044 (2002)].
Down's syndrome is caused by an extra copy of chromosome 21, the shortest human chromosome (50-million base-pairs). One birth in 700 is a Down's baby — most frequently seen in the babies of women giving birth in their 30s or 40s. The disease accounts for one-third of all cases of mental retardation in industrialized countries. Down's syndrome victims have short stature, hearing deficits and features of accelerated aging, which include hair graying & hair loss and increased tissue lipofuscin levels. One third of Down's victims have hypothyroidism. Although the overall cancer incidence may be lower, the incidence of leukemia is 10 to 20 times higher than normal. Down's syndrome victims are very vulnerable to infection, due to the rapid shortening of the telomeres of their leukocytes (white blood cells). Almost all Down's syndrome victims have Alzheimer's Disease by age 50, probably because chromosome 21 carries the amyloid gene. Chromosome 21 also carries Cu/Zn SuperOxide Dismutase gene, resulting in increased production of hydrogen peroxide (H2O2) which (without catalase or glutathione peroxidase) can lead to more hydroxyl radicals. Down's syndrome victims show a 50% increase in cytoplasmic SOD. Cultured cells transfected with an increased gene dosage of cytoplasmic SOD show features of cellular senescence mediated by hydrogen peroxide [HUMAN MOLECULAR GENETICS; de Haan,JB; 5(2):283-292 (1996)]. The incidence of diabetes is 5−10 times greater for Down's syndrome victims than for age-matched controls. Nonetheless, Down's syndrome victims show no accelerated vulnerability for breast & prostate cancer, high blood pressure, or osteoporosis.
Xeroderma pigmentosum (XP) patients show tissue-specific signs of premature aging, mainly of the skin & eyes ("photoaging"), have a high incidence of skin cancer (more than a thousand-fold over normal) and have neurological problems. Although XP victims rarely reach the age of 30, for the most part they do not display an "accelerated aging" phenotype. XP is due to compromised Nuclear Excision Repair (NER) due to defects in any one of seven genes/proteins designated XPA to XPG. XPB and XPD are helicases which are part of the NER transcription factor TFIIH. XPC and XPE are proteins that recognize DNA damage. Patients with defects in XPC (which functions exclusively in GG-NER) do not suffer the severe neurological disease that can be seen in patients with XPA defects [DNA REPAIR; Niederhofer,LJ; 7(7):1180-1189 (2008)]. XPF and XPG are endonucleases. The XPF-ERCC1 endonuclease functions in both Global Genome Nuclear Excision Repair (GG−NER) and repair of crosslinks between DNA strands. XP can even be caused by mild mutations of the XPF subunit of the XPF−ERCC1 endonuclease — which is used to replace pyrimidine dimers. DNA repair capability is particularly important in the brain because neurons are not replaced, but have high metabolic demands which subject them to high oxidative stress. For this reason, reduced repair of oxidative DNA damage is a reasonable explanation for the neurodegeneration seen in XP patients [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Reardon,JT; 94(17):9463-9468 (1997)]. Cells from XP patients with neurodegeneration show extremely poor NER of free-radical induced bulky DNA lesions [JOURNAL OF BIOLOGICAL CHEMISTRY; Brooks,PJ; 275(29):22355-22362 (2000)].
Transgenic mice which are null for both XPF & ERCC1 proteins are defective in both NER and DNA interstrand crosslink repair, which leads to XFE progeria. Defective crosslink repair leads to DSBs in these mice [MOLECULAR AND CELLULAR BIOLOGY; Niedernhofer,LJ; 24(13):5776-5787 2004)]. In XFE progeria there is more cell senescence and more apoptosis, but less mutation and telomere loss [NATURE; Niedernhofer,LJ; 444:1038-1043 (2006)]. These mice show accelerated aging, but suppressed carcinogenesis [NATURE; Niedernhofer,LJ; 444:1038-1043 (2006)]. Interstrand crosslink repair necessitates NER followed by HR [THE LANCET ONCOLOGY; McHugh,PJ; 2(8):483-490 (2001)]. Complicating matters is reduced serum IGF1, which is believed to be an adaptive response to DNA damage [NATURE; Niedernhofer,LJ; 444:1038-1043 (2006)].
Cockayne syndrome is due to a defective protein which is required for the Transcription-Coupled Repair (TCR) subtype of Nucleotide Excision Repair (NER) of DNA. Although newborns appear normal there is early dwarfism ("cachectic dwarfism"), mental retardation, cataracts, deafness, photosensitivity, osteoporosis, dental caries, sparse hair and a senile-like appearance (including a pinched, narrow face and a beaked nose, due to reduced subcutaneous fat) [PEDIATRIC NEUROLOGY; Ozdirim,E; 15(4):312-316 (1996)]. Microcephaly results from cell loss during brain development due to various kinds of DNA damage. There may be premature atherosclerosis, high lipofuscin accumulation in neurons and Alzheimer's neurofibrillary tangles. The most common causes of death are pneumonia (probably due to the general atrophy) and neurodegeneration [HUMAN MOLECULAR GENETICS; Navarro,CL; 15(2):R151-R161 (2006)]. Neurodegeneration may be indicative of the significance of failed TCR (or BER) in postmitotic tissues leading to apoptosis, and the high rate of repair required by brain tissue due to high oxidative metabolism [MECHANISMS OF AGEING AND DEVELOPMENT; Stevnsner,T; 129(7-8):441-448 (2008)]. About 25% of cases have defective CSA protein and die at an average age of 12.5, whereas the rest have defective CSB protein and die at an average age of 6.5. Oxidative stress lesions to DNA accumulate at a rapid rate in victims with CSB defects, indicative of deficient Base Excision Repair (BER) [THE FASEB JOURNAL; Tuo,J; 17(6):668-674 (2003) and MOLECULAR AND CELLULAR BIOLOGY; de Waard,H; 24(18):7941-7948 (2004)]. Despite showing many of the same symptoms as XP victims, Cockayne syndrome victims have no predisposition to cancer because of inhibited transcription binding leading to a high rate of apoptosis (to which is attributed features of premature aging). Cancer cells are heavily dependent upon transcription, so it is reasonable that defective TCR would strongly inhibit proliferation, but favor high levels of senescence or apoptosis ("accelerated aging" rather than cancer) [THE AMERICAN JOURNAL OF HUMAN GENETICS; Licht,CL; 73(6):1217-1239 (2003)]. Transgenic mice that replicate Cockayne Syndrome show suppression of GH/IGF1−1PLoS BIOLOGY; van der Pluijm,I; 5(1):e2 (2007)].
Trichothiodystrophy (TTD) is due to defects in the transcription factor TFIIH protein required for both NER and normal transcription. Most often these defects are in the XPB (3´−>5´ helicase) or XPD (5´−>3´ helicase) subunits of TFIIH. One type of mutation in XPD leads to xeroderma pigmentosum, whereas other mutations lead to TTD [HUMAN MOLECULAR GENETICS; Botta,E; 11(23):2919-2928 (2002)]. TTD patients do not show increased incidence of cancer. Whereas XP patients experience neuronal degeneration, Cockayne Syndrome (CS) and TTD patients suffer from failure to develop brain myelin. CS and TTD patients may suffer more from defective normal transcription, whereas XP patients may suffer more from defective transcription in NER [NEUROSCIENCE; Kraemer,KH; 145(4):1388-1396 (2007)]. TTD victims (including transgenic mice) show more accelerated aging than victims of Werner, Cockayne or Bloom Syndrome. The aging phenotype (which includes osteoporosis, early greying, cachexia and neurological abnormalities) is attributed to increased apoptosis as well as impaired cell functioning [SCIENCE; de Boer,J; 296:1276-1279 (2002)].
Like XP, Ataxia Telangiectasia (AT) is a hereditary disease (defective Ataxia Telangiectasia Mutated, ATM, gene) that reduces DNA repair and greatly increases the risk of cancer. Unlike XP, AT affects DSB repair rather than NER, and mainly increases leukemia or lymphoma cancer-types. AT victims exhibit growth retardation, gonadal atrophy, graying hair, immune deficiency, accelerated telomere loss, genetic instability and cerebellar degeneration, particularly of Purkinje and granule neurons. Neurodegeneration is the most prominent feature of AT [NEUROMOLECULAR MEDICINE; Frappart,P; 8(4):495-511 (2006)] as a result of the vulnerability of Purkinje cells to oxidative stress [THE JOURNAL OF NEUROSCIENCE; Chen,P; 23(36):11453-11460 (2003)]. "Ataxia" means impaired motor coordination, and ATM victims require a wheelchair before becoming teenagers due to loss of cells in cerebellum. Clusters of dilated blood vessels (telangiectasia, "spider veins") appear on the whites of eyes. ATM victims usually die in their teens [CELL; Rass,U; 130(6):991-1004 (2007)]. Inhibition of ATM under conditions of DNA damage reduces cell senescence and increases apoptosis [JOURNAL OF BIOLOGICAL CHEMISTRY; Zhang,X; 280(20):19635-19640 (2005)] or simply reduces senescenceMOLECULAR BIOLOGY OF THE CELL; Moiseeva,O; 17(4):1583-1592 (2006)]. Nonetheless, cells of AT patients are resistant to apoptosis and do not undergo cell cycle arrest when subjected to ionizing radiation, possibly due to the anti-apoptotic action of AP−1 [JOURNAL OF BIOLOGICAL CHEMISTRY; Weizman,N; 278(9):6741-6747 (2003)]. Tissues containing rapidly-dividing cells (such as the cells in the epithelium) show most of the signs of aging. But the normal slow loss of cerebellar Purkinje cells is greatly accelerated, leading to ataxia. Ca2+/cAMP Response Element Binding protein CREB has anti-apoptotic action related to transcription of Bcl−2 and neuron growth factors. Dysregulation of ATM-mediated phosphorylation of CREB in response to DNA damage may be a significant factor in the neurodegeneration associated with AT [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Shi,Y; 101(6):5898-5903 (2004)]. Although the primary symptom of AT is degeneration of the cerebellum, the ATM protein is activated by DNA double-strand breaks [SCIENCE; Abraham, RT; 308(5721):510-511 (2005)]. ATM stimulation in AT patients is more sensitive to ionizing radiation & alkylating agents than to ultraviolet radiation, whereas the reverse is true in XP patients. XP patients are defective in NER, whereas AT patients are defective in cell cycle control [JOURNAL OF RADIATION RESEARCH; Kan'o,M; 48(1):31-38 (2007)]. Additionally, absence of ATM protein increases telomerase instability & loss of telomeres [ONCOGENE 21:611-618 (2002)] because DNA-damage response is part of telomere protection and maintenance [NATURE; Verdun,RE; 447:924-931 (2007)].
Like XP, Fanconi Anemia (FA) is a hereditary disease associated with chromosome instability and greatly increased risk of cancer. Increased apoptosis of hematopoetic cells typically leads to pancytopenia by age 7, with the surviving cells characterized by genetic instability leading to acute myeloid leukemia (AML, the most common cause of cancer in FA patients) [CELL;Niedernhofer,L; 123(7):1191-1198 (2005) and NATURE REVIEWS; D'Andrea,AD; 3(1):23-34 (2003). Median age of death is 16, most commonly due to bone marrow failure. Premature aging features include progressive bone marrow failure, premature reproductive aging, hyperinsulinaemia, hypothyroidism and growth hormone deficiency [NUCLEIC ACIDS RESEARCH; Grillari,J; 35(22):7566-7576 (2007)]. Other FA symptoms are only marginally associated with an accelerated aging phenotype: hearing impairment, skeletal abnormalities, cardiac abnormalities, and cafe-au-lait skin spots. All cellular elements in the blood (erythrocytes, leukocytes, and platelets) are depressed. FA symptoms can result in disruption of proteins required for repair of interstrand DNA cross-links, inclucing BRCA2. Replication fork arrest during the S phase of the cell cycle due to DNA cross-links can activate FA protein complexes to excise cross-links and thereby create DSBs that can be repaired by homologous recombination [GENES & DEVELOPMENT; Kennedy,RD; 19(24):2925-2940 (2005)]. An increase in anemia prevalence is regarded as part of the normal aging phenotype [JOURNAL OF THE AMERICAN GERIATRIC SOCIETY; Rothstein,G; 51(3 Suppl):522-526 (2003)], and spontaneous DNA interstrand crosslink damage has been proposed to be the cause of that anemia [THE EMBO JOURNAL; Prasher,JM; 24(4):861-871 (2005)].
An analysis of human segmental progerias observed that the progerias with shortened telomeres also showed grey hair, alopecia and nail atrophy — whereas those not having shortened telomeres did not have those features. The study even hypothesized that fingernail growth velosity may be a biomarker of aging [JOURNAL OF GERONTOLOGY; Hofer,AC; 60(1):B10-B20 (2005)]. A similar study observed that although most segmental progerias are associated with increased risk of cancer, Hutchinson-Gilford progeria and Cockayne syndrome are not. The study concluded that lipid metabolism genes are more influential on human lifespan than genome integrity genes [THE INTERNATIONAL JOURNAL OF BIOCHEMISTRY & CELL BIOLOGY; 37(5):947-960 (2005)].
The Senescence Accelerated Mouse (SAM) is a rodent model of accelerated aging which is apparently related to free-radical damage, judging by various indices of such damage in the rodent. Moreover, administration of the spin-trapping agent PBN at maturity to reduce free-radical damage dramatically increases life span — providing support for the free radical theory of aging [ANNALS OF THE NEW YORK ACADEMY OF SCIENCES 854:239-250 (1998)]. This is in contrast to other segmental progerias which more often support a DNA-repair-deficiency theory of aging.
Mouse studies indicate that mutants for Ku80/Ku86 — proteins essential for NHEJ — show an accelerated aging phenotype (kyphosis, alopecia, conjunctivitis, rectal prolapse, osteopenia, skin atrophy, epiphysial closure, reduced lifespan), but a 13-fold reduction of cancer incidence [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Vogel,H; 96(19):10770-10775 (1999) and MOLECULAR AND CELLULAR BIOLOGY; Li,H; 27(23):8205-8214 (2007)]. The control mice showed osteoporosis, epiphysial closure as well as skin and follicular atrophy after 70 weeks or greater, whereas these symptoms were seen in the Ku 86 knockout mice at 37 weeks, 22 weeks, and 37 weeks, respectively [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Vogel,H; 96(19):10770-10775 (1999)]. The Ku 86 knockout mice exhibited earlier onset of cancer, despite an overall reduced incidence (which may have been due to the shortened lifespan and the increased cellular senescence [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Vogel,H; 96(19):10770-10775 (1999)]. Mice with mutated XPD (a protein required for the TFIIH protein that causes DNA unwinding in NER) show an accelerated aging phenotype which includes osteoporosis, kyphosis, early graying, cachexia and reduced lifespan [SCIENCE; de Boer,J; 296:1276-1279 (2002)]. Mice mutant for reduced levels of Brca1 (a protein for DSB repair) show both increased cellular senescence and an accelerated aging phenotype (kyphosis, osteoporosis, slow wound healing, reduced dermal thickness, muscular atrophy) [GENES & DEVELOPMENT; Cao,L; 17(2):201-213 (2003)]. Mice with deficient p53-related p63 protein also show increased cellular senescence associated with tissue histology reflecting an accelerated aging phenotype [GENES & DEVELOPMENT; Keyes,WM; 19(17):1986-1999 (2005)]. Similarly, mice deficient in ZMPSTE24 protease show hyperactivation of tumor suppressor protein p53, leading to increased cell senescence without increased apoptosis — and an acclerated aging phenotype [NATURE; Varela,I; 427:564-568 (2005)].
The linkage of accelerated aging to nuclear DNA repair defects implies both a direct linkage to cancer as well as cell dysfunction due to increased DNA damage/mutation, or an indirect linkage due to increased cellular senescene and apoptosis — depending on what causes the "aging phenotype". Transgenic mice with hyperactive p53 protein show decreased cancer along with increased apoptosis and cellular senescence associated with an aged phenotype and shortened lifespan [NATURE; Tyner,SD; 415:45-53 (2002), GENES & DEVELOPMENT; Maier, B; 18(3):306-319 2004), and BLOOD; Dumble,M; 109(4):1736-1742 (2007)]. Markers of aging for the first of these studies included hair sparseness (hair growth decreases linearly with age in mice), slowing of wound healing, reduced dermal thickness & subcutaneous adipose (both of which normally decline with age), lordokyphosis (hunchbacked spine), muscle atrophy, and reduced vigor [NATURE; Tyner,SD; 415:45-53 (2002)]. Conversely, transgenic mice with mutated p66shc gene show impaired p53, reduced apoptosis in response to stress and "decelerated aging" (lifespan extended 30% [NATURE; Migliaccio,E; 402:309-313 (1999)]. Transgenic mice with extra p53 genes had normal basal p53 activity, normal lifespans (no signs of accelerated aging), and enhanced resistance to DNA damage and cancer — probably because of enhanced protection against p53 mutation [THE EMBO JOURNAL; Garcia,CI; 21(22):6225-6235 (2002)]. Mice that were transgenic with extra genes of both p53 and Arf (with normal activity of both) had strong cancer resistance, increased levels of reduced glutathione (GSH) antioxidant, and lifespan increase of 16% [NATURE; Matheu,A; 448:375-380 (2007)].
In the liver (but not the brain), old rats show less than half the apoptosis of young rats in response to DNA damage [NATURE MEDICINE; Suh,Y; 8(1):3-4 (2002)], indicative of an increased vulnerability to cancer. Cellular senescence isn't simply a result of shortened telomeres, it is often the result of unrepaired nuclear DNA damage throughout chromosomes [NATURE CELL BIOLOGY; Sedelnikova,OA; 6(2):168-170 (2004) and MECHANISMS OF AGEING AND DEVEOPMENT; von Zglinicki,T; 126(1):111-117 (2005)], although telomere-initiated senescence is probably also a DNA damage response [NATURE; d'Adda di Fagagna,F; 426:194-198 (2003)]. Although estimates of the number of senescent cells vary from between less than 1% to over 15%, such cells are prominent in osteoarthritis and atherosclerosis — and, where not prominent, contribute to cellular dysfunction and carcinogenesis of adjacent cells by secretion of cytokines, growth factors and other damaging agents [NATURE REVIEWS; Campisis,J; 8(9):729-740 (2007) and JOURNAL OF BIOLOGICAL CHEMISTRY; Coppe,J; 281(40):29568-29574 (2006)]. Foci of DNA damage as markers of senescent cells provide the highest estimates (15%) of cellular senescence in aged animals [SCIENCE; Herbig,U; 311:1257 (2006)].
Accelerated aging diseases can be useful models for learning about the mechanisms of aging if they truly represent accelerated aging. When the question of the essence of aging remains undetermined, validating biomarkers or a model of accelerated aging leads to circular reasoning. Some question that there are any disease conditions which can be called "accelerated aging" [AGING CELL; Miller,RA; 3(2):47-51 (2004)]. If it is possible to slow aging, it should be possible to accelerate aging, but proving that aging has been slowed is much easier than proving that aging has been accelerated because a long-lived organism is sufficient proof of decelerated aging but a short-lived organism could be the result of a specific defect. Or else the mechanisms of "accelerated aging" could be different from those of "normal aging". But lifespan studies of nematodes show progressive lamin disorganization in normal aging comparable to that in the accelerated aging of HGPS progeria, and the rate of these changes can be manipulated by insulin/IGF−1-like signaling [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Haithcock,E; 102(46):16690-16695 (2005)]. Similar defects in nuclear structure & function between HGPS cells and cells in elderly humans also supports the contention that HGPS is accelerated aging [SCIENCE; Scaffidi,P; 312:1059-1063 (2006)].
Dermatologists commonly distinguish photoaging from chronological aging in the skin, attributing most skin aging to the former rather than the latter. Singlet oxygen from ultraviolet light increases mitochondrial DNA deletion [JOURNAL OF BIOLOGICAL CHEMISTRY; Berneburg,M; 274(22):15345-15349 (1999)]. Ultraviolet radiation stimulates collagen degradation while inhibiting collagen production [AMERICAN JOURNAL OF PATHOLOGY; Quan,T; 165(3):741-751 (2004)], increases oxidative DNA damage [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Kozman,S; 102(38):13538-13543 (2005)] and causes stress-induced premature senescence in skin fibroblasts [JOURNAL OF CELL SCIENCE; Debacq-Chainiaux,F; 118(Pt 4):743-758 (2005)]. If aging is damage to macromolecules, cells and tissues then it should not be surprising that exogenous sources of damage could cause "accelerated aging" of specific tissues — a "tissue-specific segmental progeria". The fact that nuclear DNA damage from exogenous agents results in a phenotype that greatly resembles normal aging lends credence to the idea that such damage is the basis of normal aging.
Mice have been regarded as examples of "accelerated aging" [AGING CELL; Miller,RA; 3(2):47-51 (2004)], but mice could also be given as examples of segmental human progeria because of their high susceptibility to cancer and their absence of atherosclerosis and Alzheimer's Disease. All human aging could be called segmental in the sense that some people get cancer, atherosclerosis and Alzheimer's Disease, whereas others do not. If there are multiple forms of aging damage then there cannot be a single aging biomarker or aging phenotype. If reduced longevity alone were a sufficient criterion for accelerated aging, then any genetic defect which increases mortality — or even a dangerous occupation — would qualify.
The nematode worm Caenorhabditis elegans (C. elegans, which is the size of a comma, and lives a few weeks) and the fruit fly Drosophila melanogaster (Drosophila, which lives a few months) are the most common invertebrate model species used in biology. Upon reaching maturity both species are composed post-mitotic cells, except for the germline.
A study of the entire genome of Drosophila during the aging process has led to the conclusion that 300 to 350 genes control aging [CURRENT BIOLOGY; Pletcher,SD; 12(9):712-723 (2002) and CURRENT BIOLOGY; Rose, MR; 12(9):R311-R312 (2002)]. (By extrapolation, about 500 genes would control aging in humans.) Determining what those genes do would be a major step toward understanding the causes of aging.
Drosophila have the natural antioxidant enzymes SuperOxide Dismutase (SOD) & CATalase (CAT), but no glutathione peroxidase. Drosophila were created with (1) extra Cu/Zn−SOD (cytoplasmic SOD) genes (2) extra CAT genes and (3) extra Cu/Zn−SOD and extra CAT genes. Only the flies in the third category, having both extra genes showed extended lifespan. These transgenic flies showed 26% greater SOD activity, 73% greater CAT activity and 34% longer lifespan [JOURNAL OF BIOLOGICAL CHEMISTRY 270(26):15671-15674 (1995)]. The authors later stated that the experiments were not conclusive because of the genetic background of the organisms and because of artifacts of the transgenic methods [EXPERIMENTAL GERONTOLOGY; Orr,WC; 38:227-230 (2003)]. Nonetheless, transgenic mice expressing mitochondrial catalase by 50 times that seen in normal mice increased maximum lifespan by about 20% [SCIENCE; Schriner,SE; 308:1909-1911 (2005)].
In 1988 geneticist Tom Johnson of the University of Colorado announced the discovery of a mutant gene in C. elegans that at 25ºC increased mean life span 65% and maximum lifespan 110% [GENETICS 118:75-86 (1988)]. Johnson named the gene age−1 in the expectation that other genes for aging would be found. Caloric Restriction with Adequate Nutrition (CRAN) further extends the lifespan of age−1 mutants. age−1 mutants were shown to have elevated Cu/Zn SOD and CAT (nematodes, unlike vertebrates, do not have glutathione peroxidase) [BIOCHEMICAL JOURNAL 292:605-608 (1993)]. age−1 mutants show a lower rate of deletions in the mitochondrial genome than wild-types [NUCLEIC ACID RESEARCH 23(8):1419-1425 (1995)]. C. elegans homozygous for nonsense age−1 gene mutations have shown a ten-fold increase in maximum lifespan [AGING CELL; Ayyadevara,S; 7(1):13-22 (2008)].
Nematodes normally live under the soil where oxygen concentrations are 1% to 2%, suggesting that the higher levels of antioxidant enzymes may only be of advantage as an adaptation to the atmospheric oxygen (21% oxygen) of laboratory conditions. When age−1 mutants are raised under more natural conditions of low food-availability they die more quickly than wild-type [NATURE 405:296-297 (2000)].
C. elegans age−1 was later identified [GENETICS; Malone,EA; 143(3):1193-1205 (1996)]. as daf−23, part of the gene family including the daf−2 (DAuer Formation gene) mutation of which causes the nematode to go into the developmentally arrested dauer state (from the German dauern, meaning "to endure"). The daf−2 DNA gene sequence most resembles the mammalian gene for the IGF1−1 (Insulin-like Growth Factor−1) receptor, but is also quite similar to the insulin receptor. (In C. elegans the single daf−2 receptor corresponds in function to the two mammalian receptors — insulin and IGF−1.) The age−1 gene product is a signaling kinase which acts downstream of the daf−2 receptor.
The dauer state is naturally seen among C. elegans larvae under conditions of low food availability. The dauer is non-feeding, non-reproductive and resistant to damage from ionizing radiation, extreme temperature & free-radicals ("stress-resistant"). If conditions improve, the dauer moults to a normal adult state. Autophagy genes are essential for dauer formation and are essential for the lifespan increase associated with dauer [SCIENCE; Melendez,A; 301:1387-1391 (2003)].
Dauer larvae do not feed and the time spent in a quiescent state of reduced metabolism ("suspended animation") may not count as life extension. Some ticks stop metabolizing if they cannot eat, and can survive in a quiescent state for years. Drosophila cooled from 25ºC to 15ºC live more than 3 times longer [JOURNAL OF BIOLOGICAL CHEMISTRY; 32(1):103-121 (1917)] — a temperature reduction which should be associated with a halving of metabolic rate.
Among other effects, Daf proteins reduce fertility & movement of C. elegans while shifting metabolism toward the breakdown of fat — analogous to the metabolic shift seen in humans when insulin levels fall off. But the lifespan increases of the dauer state are not entirely due to reduced metabolism — increased antioxidant enzyme levels and more stress-resistance proteins play a role. Strong reduction of function mutations to daf−2 or age−1 (the "insulin/IGF−1 pathway") cause the dauer state, whereas weak mutations simply cause a more quiescent phenotype having dauer-like qualities of increased antioxidant enzymes, extended lifespan and lower metabolic rate [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); van Voorhies,WA; 96(20):11399-11403 (1999)]. In particular, daf−2 mutations increase Mn−SOD expression in the mitochondria [THE FASEB JOURNAL; Honda,Y; 13(11):1385-1393 (1999)].
Not only are daf−2 mutations associated with increased catalase, but C. elegans with defective peroxisomal catalase genes have been proposed as models of "accelerated aging" [JOURNAL OF BIOLOGICAL CHEMISTRY; Petriv,OI; 279(19):19996-20001 (2004)]. Some daf−2 mutants display the same phenotype, but without reduced fertility [GENETICS; Gems,D; 150(1):129-155 (1998)]. Removal of gonads from daf−2 reduction-of-function nematodes increased lifespan even more than the reduction of daf−2 function [SCIENCE; Arantes-Oliveira,N; 302:611 (2003)]. C. elegans on CRAN get further life extension with daf−2 mutations, indicating that CRAN and insulin/IGF−1 defects extend lifespan by different mechanisms [EXPERIMENTAL GERONTOLOGY; Houthoofd,K; 38(9):947-954 (2003) and PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Lakowski,B; 95(22):13091-13096 (1998)].
Reduced expression of daf−2 or age−1 cannot induce the dauer state or extend lifespan without the Daf−16 transcription factor, which is downstream from the other two proteins. The signalling dependency is:
daf−2 -> age−1 -> PKB -> Daf−16
where PKB is Protein Kinase B. Note that the dependency relationship is such that high "insulin signalling" (high daf−2/age−1 expression) leads to low Daf−16 activity, whereas defective mutations of daf−2/age−1 (or starvation) leads to high Daf−16 transcription activity, which causes the dauer-like lifespan extension.
A table summarizes the homologous proteins between C. elegans and mammals:
| C. elegans | daf−2 | age−1 | PKB | Daf−16 | Daf−18 | |
| mammals | Ins/IGF−1 | PI3K | Akt | FOXO | PTEN |
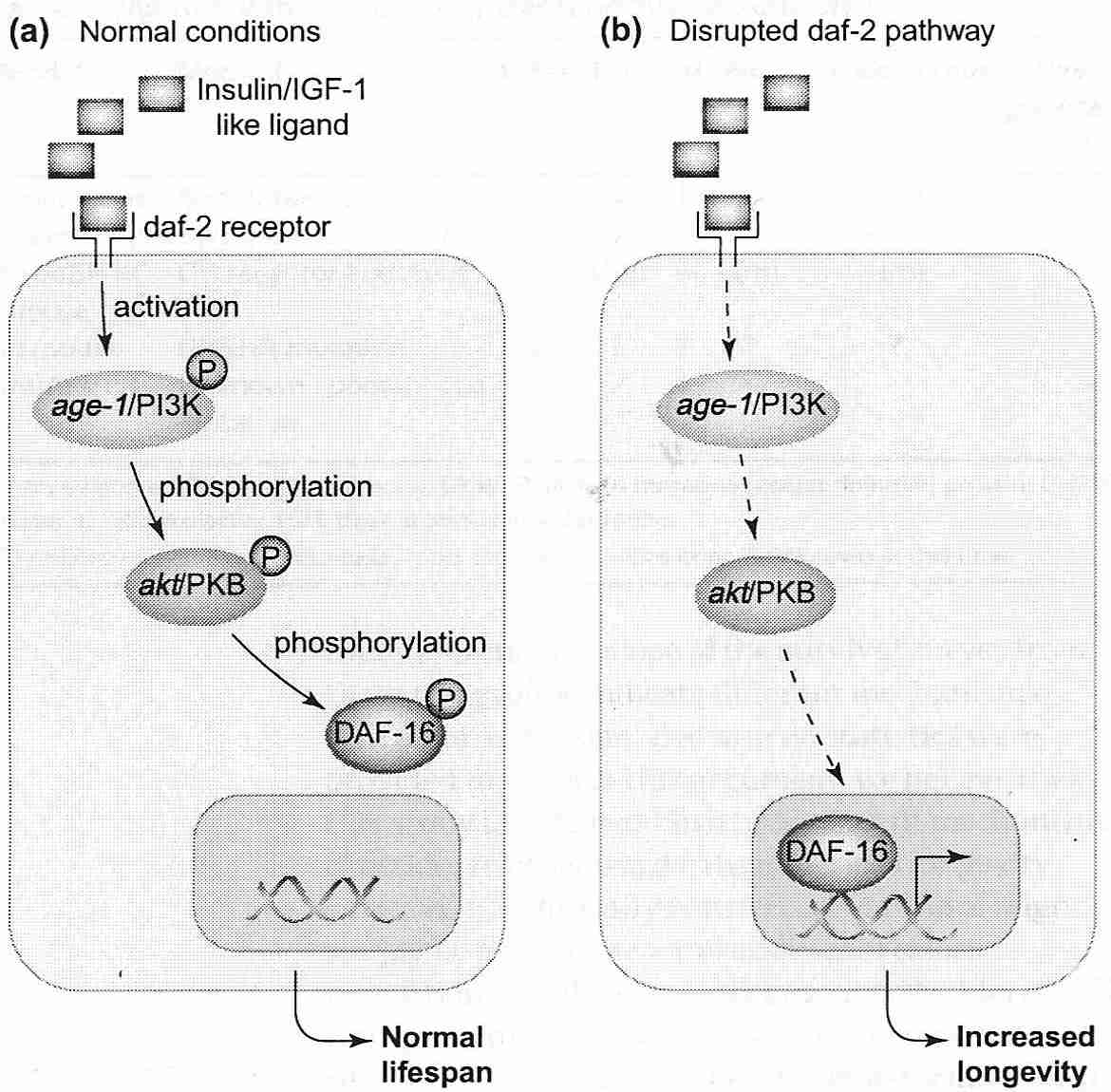
|
Mammalian homologs to the C. elegans genes/proteins are the subject of intense interest concerning possible regulation of mammalian lifespan. As has been mentioned, the C. elegans daf−2 receptor acts like a mammalian insulin/IGF−1 receptor. C. elegans age−1 corresponds in mammals to the inositol lipid kinase PI3K (PhosphatidylInositol 3−Kinase). Mammalian protein kinase B (PKB) is also called Akt (a kinase activated by phosphorylation of serine and/or threonine residues). Daf−16 corresponds to the mammalian family of transcription factors called FOXO (Forkhead bOX class O), which regulates stress response. When FOXO proteins are phosphorylated by protein kinase B they are excluded from the nucleus and degraded upon ubiquitination. Daf−16/FOXO gene transcription can lead to DNA-repair stimulation, (cell cycle arrest, apoptosis, and induction of heat shock proteins & anti-oxidant enzymes [SCIENCE; Brunet,A; 303:2011-2015 (2004)]. Variations in FOXO genetics has a significant effect on human lifespan [EUROPEAN JOURNAL OF HUMAN GENETICS; Kuningas,M; 15(3):294-301 (2007)]. Stress signals from JNK MAPKs exert their effects through Daf−16/FOXO in parallel with insulin/IGF−1 signalling [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Oh,S; 102(12):4494-4499 (2005)].
In sum, when the daf−2/insulin/IGF−1 pathway is intact, age−1/PI3K phosphorylation of Daf−16/FOXO prevents that protein from entering the nucleus to activate defensive/hibernation "longevity genes".
The mammalian PI3K/Akt pathway activates cell growth & proliferation while at the same time promoting cell survival by inhibiting macroautophagy and apoptosis [JOURNAL OF BIOLOGICAL CHEMISTRY; Arico,S; 276(38):35243-35246 (2001)]. PTEN normally dampens the PI3K/Akt pathway, thereby acting as a tumor-suppressor. By inactivation of the tumor suppressor protein PTEN, oxidative stress activates PI3K — resulting in PKB/Akt promotion of proliferation [THE EMBO JOURNAL; Leslie,NR; 22(20):5501-5510 (2003)]. PI3K activation increases expression of ARE (Antioxidant-Responsiveness Element) genes leading to the synthesis of more antioxidant enzymes. PTEN mutations that inactivate PTEN expression allow unrestrained activity of PI3K/Akt, which often leads to cancer [MEDICAL SCIENCE MONITOR; Chu,EC; 10(10):RA235-RA241 (2004)]. The mammalian PTEN gene corresponds to the C. elegans daf−18 gene. Mutations in daf−18 can suppress the dauer phenotype and longevity induced by daf−2 or age−1 inactivation [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Mihaylova,VT; 96(13):7427-7432 (1999)].
(Human atherosclerotic plaques contain a high proportion of senescent cells, probably because of increased p53-induced senescence as a result of Akt phosphorylation by insulin — underlying the relationiship between diabetes and atherosclerosis. These senescent cells produce high levels of proinflammatory molecules that promote atherogenesis. Inhibition of FOXO3 by Akt is an essential factor in the senescent growth arrest [THE EMBO JOURNAL; Miyauchi,H; 23(1):212-220 (2004)].)
The mammalian PI3K/Akt pathway can be inhibited by rapamycin, which inhibits the kinase mTOR/TOR — (mammalian) Target Of Rapamycin. Inhibition of mTOR by rapamycin or calorie restriction triggers macrophagy. Drosophila fed rapamycin have shown extended lifespan [CELL METABOLISM; Bjedov,Z; 11(1):35-46 (2010)] and rapamycin has the same effect on mice [NATURE; Harrison,DE; 460:392-396 (2009)]. Changes in the expression of genes controlled by Tor, Ras, and Sch9 in yeast causes a switch to glycerol calorie source — extending lifespan [PLOS GENETICS; Wei,M; 5(5):e1000467 (2009)]. (For more information on the PI3K/Akt pathway see Signalling Molecules and Transcription Factors.)
In Drosophila a defective chico "insulin/IGF−1 signalling gene" has been shown to increase lifespan 48%, reduce fertility and increase antioxidant enzymes and produce a fly that is half the size of wild-type flies [SCIENCE; Clancy,DJ; 292:104-106 (2001)]. Chico mutants have fewer cells and smaller cells [CELL; Bohni,R; 97(7):865-875 (1999)]. As with C. elegans, the single insulin/IGF−1 receptor in Drosophila is believed to correspond with the distinct (but similar) insulin and IGF−1 receptors in humans. Chico protein corresponds to the mammalian Insulin Receptor Substrate (IRS) "docking protein" that is associated with the IGF−1 receptor, so defective mutations result in a similar effect as defective daf−2/age−1/Insulin/IGF−1 signalling.
The C. elegans gene clk−1 (the "clock" gene), alters growth rate, cell cycle time and other "timed" events in the nematode life-cycle. Defective clk−1 genes reduce metabolism and extend lifespan, whereas overexpression of clk−1 reduces lifespan. The sluggish clk−1 mutants are defective in Coenzyme Q synthesis (essential for mitochondrial energy generation) and when they are fed bacteria that do not supply CoEnzyme−Q their development arrests [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA) 98(2):421-426 (2001)], and they revert to anaerobic metabolism that produces less energy while avoiding much of the free radical generation associated with oxidative phosphorylation [SCIENCE; Larsen, PL; 295:120-123 (2002)].
C. elegans lifespan extension has been achieved by mitochondrial function inhibition [SCIENCE;Dillin,A; 298:2398-2401 (2002)]. C.elegans given anti-oxidant compounds that mimic the action of both superoxide dismutase and catalase show significant increases in mean and maximum lifespan [SCIENCE; Melov,S; 289:1567-1569 (2000)]. Nematode daf−2/age−1 impairment only increases lifespan when the defect occurs in neurons, not muscle or intestine — possibly because neurons are more vulnerable to free-radicals [SCIENCE; Wilkow,CA; 290:147-150 (2000)]. The HSF−1 transcription factor (Heat Shock Factor, which regulates heat shock response) has been shown to be essential for Daf−16 induced longevity in C.elegans. Reduced activity of HSF−1 reduces lifespan and additional HSF−1 gene copies have increased lifespan by 40%. Small heat shock proteins which can inhibit toxic protein aggregation seem to be the key to this effect [SCIENCE; Hsu,A; 300:1142-1145 (2003)].
Similarly, Drosophila bred for longevity have displayed increased levels of small heat shock proteins [JOURNALS OF GERONTOLOGY; Kurapati,R; 55A(11):B552-B559 (2000)]. Overexpression of small heat shock proteins in Drosophila has been shown to extend their lifespan by 30% [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Wang,H; 101(34): 12610-12615(2004)].
A number of mutations can produce the effects of CRAN by indirect means. For example, the eat mutations in C. elegans result in reduced efficiency of the pharynx to pump food [GENETICS; McKay,JP; 166(1):161-169 (2004)]. More subtly, the Indy (I'm Not Dead Yet) mutation in Drosophila results in defective membrane transport of Krebs cycle intermediates [SCIENCE; Rogina,B; 290:2137-2140 (2000)].
Metabolic rate is not reduced in C. elegans daf−2 mutants having extended lifespan, but efficiency of oxidative phosphorylation is increased [AGING CELL; Braeckman,BP; 1(2):82-88 (2002)]. Nor is metabolic rate reduced in Drosophila, either in CRAN or defective insulin/IGF−1 signalling (chico mutants) having extended lifespan [EXPERIMENTAL GERONTOLOGY; Hulbert,AJ; 39(8):1137-1143 (2004)]. Cohorts of Drosophila with no obvious genetic defects can have up to a five-fold difference in lifespan with no significant difference in metabolic rate [EXPERIMENTAL GERONTOLOGY; Hulbert,AJ; 39(8):1137-1143 (2004) and JOURNAL OF APPLIED PHYSIOLOGY; van Voorhies,WA; 95(6):2605-2613 (2003)].
Mice (and other mammals) have distinct insulin and IGF1−1 receptors, unlike flies and worms, that have a single insulin/IGF−1-like receptor. But defects of either of these receptors have been shown to result in a lifespan increase for mice. Insulin resistance is associated with diabetes and is even recommended as a biomarker of aging — so it is mysterious why blocked insulin signaling can extend lifespan. Fat-specific Insulin Receptor Knock-Out (FIRKO) mice have reduced fat mass, normal calorie intake and an increased maximum lifespan of 18% [SCIENCE; Bluher,M; 299:572-574 (2003)]. Yet deletion of all insulin receptor genes in mice results in neonatal death [EMBO JOURNAL; Joshi,RL; 15(7):1542-1547 (1996)].
Two single gene mutations on mice — one on chromosome 11 (Prop−1 locus, Ames dwarf) and the other on chromosome 16 (Pit−1 locus, Snell dwarf) — extend mean & maximum lifespan significantly. Snell dwarfs have a defective pituitary transcription factor which is downstream from the protein which is defective in Ames dwarfs. Both mutations preclude normal development of the anterior pituitary. In both cases the adults are one-third the size of normals and in both cases there are defects in production of Growth Hormone (GH), prolactin and Thyroid-Stimulating Hormone (TSH). Comparable to nematode daf−2/age−1 mutants, dwarf mice have impaired IGF−1/insulin sensing pathways.
Insulin-like Growth Factor−1 (IGF−1) is a mitogen and an important mediator of the GH effect. The dwarf mice have greatly diminished IGF−1 blood levels. Dwarf mice have higher antioxidant enzyme activity, lower body temperature and reduced metabolism [JOURNALS OF GERONTOLOGY 56A(8):B340-B349 (2001)] as well as delayed collagen cross-linking and delayed immune (T−cell) aging [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Flurkey,K; 98(12):6736-6741 (2001)]. Fibroblasts from Snell Dwarf mice have great resistance to injury by heat, hydrogen peroxide, cadmium, UV light and paraquat [FASEB JOURNAL 17:1565-1566 (2003)] — a similar stress-resistant pattern as is seen in daf−2/age−1 mutants. Kidney disease is common in rodents, but this is not the case for dwarfs.
The slope of the survival curves of dwarf mice match those of controls, indicating that the rate of aging is not changed — rather the curve has been shifted to the right, possibly due to slower development time to maturity. Ames dwarfs develop tumors at the same frequency as wild-types, but at a later age [JOURNAL OF THE AMERICAN AGING ASSOCIATION; Mattison,JA; 23:9-16 (2000)]. CRAN, by contrast, reduces the slope of the survival curve. The fact that the lifespan of Ames Dwarf mice can be further extended by CRAN [NATURE; Barke,A; 414:412 (2001)] shows that the mechanism of lifespan extension in dwarfism is at least partially distinct from that of CRAN. Ames Dwarf mice show a significantly delayed occurrence of cancer compared to normal mice [JOURNALS OF GERONTOLOGY; Ikeno,Y; 58A(4):B291-B296 (2003)] — probably because IGF−1 promotes apoptosis in unanchored cells (notably, cancer cells) and is anti-apoptotic in other cells.
"Knockout mice" (ie, mice with genes "knocked out") lacking GH show significantly reduced IGF−1 and thyroid hormone. The knockout mice are one-third normal weight and show a 60% lifespan extension (comparable to those of dwarf mice) [ENDOCRINOLOGY; Coschigano,KT; 141(7):2608-2613 (2000)]. As with Ames dwarfs, insulin sensitivity is greater and plasma glucose & insulin may be reduced, resulting in less glycation [JOURNALS OF GERONTOLOGY; 56A(8):B340-B349 (2001)]. Although knockout mice for IGF−1 are not viable, mice with half their IGF−1 genes knocked-out (heterozygous knockouts) live 26% longer and show resistance to oxidative stress without dwarfism, altered fertility or altered metabolism [NATURE; Holzenberger,M; 42:182-187 (2003)].
Mice with defective p66shc gene resist apoptosis caused by paraquat, hydrogen peroxide and UV light. The mice show a 30% increase in lifespan (less than the lifespan increase in dwarf mice [NATURE; Migliaccio,E; 402:309-313 (1999)]. The p66shc signal transduction pathway is activated by oxidative stress and leads to apoptosis. Apoptosis due to oxidative stress is mediated by p53, and antagonized by p21 (presumably sometimes by a p53-independent pathway in which p66shc participates). The p66shc protein increases cellular oxidative stress as a defense against infectious agents (lab mice are protected from pathogens). The p66shc mutant mice do not survive well in cold (lab mice are kept warm). The p66shc protein is downstream from the IGF−1 receptor and is underphosphorylated in dwarf mice [HORMONE RESEARCH; Hozenberger,M; 62(Suppl 1):89-92 (2004)]. The protein is an activator of the Ras mitogen receptor.
Overexpression of the klotho gene extends the maximum lifespan of male mice about 20%, but has little effect on females. Males have reduced insulin sensitivity, but females do not. Despite resistance to both insulin & IGF−1, the mice have normal size & food intake, but fertility is reduced [SCIENCE; Kurosu,H; 309:1829-1833 (2005)].
Reduced body size within a species often correlates with longer lifespan and reduced plasma IGF−1. Great Danes (400 ng/mL plasma IGF−1) live about 7 years, whereas Chihuahuas (40 ng/mL plasma IGF−1) can live over 15 years. Does low IGF−1 causing small body size lead to greater longevity — or is the smaller body size due to reduced IGF−1 irrelevant to life-extension resulting from IGF−1 signalling? Ironically, GH (and therefore, IGF−1) hormone replacement is touted as an anti-aging, rejuvenating remedy for older humans — including such claimed benefits as improved cognition & improved immune function (benefits attributed to reduced IGF−1 in mice).
It may be that high GH/IGF−1 is an example of "antagonistic pleiotrophy". Larger animals with greater fertility and short lifespans are likely to be dominant in an environment where successful competition with other animals is the key to survival. In environments with scarce resources, but little competition, smaller size with reduced fertility & greater longevity may result in more surviving offspring.
High GH and IGF−1 increases tissue development, metabolism and glucose utilization at the cost of higher oxidative stress, more protein glycation and higher proliferation. It may well be most conducive for survival to have high GH/IGF−1 during development, but reduced GH/IGF−1 levels after maturation. From this point of view, GH replacement in adults may not be a good idea.
Within mammalian species, small size is associated with greater longevity when the small size is not due to inadequate nutrition. If DNA repair capability increases for larger species, but is the same for larger members of the same species (who have more cells), then the larger animals may be more vulnerable to tissue degeneration and cancer than the smaller members of the same species. Dwarf mice and smaller breeds of dogs have less Insulin-like Growth Factor−1 (IGF−1) and are less vulnerable to cancer [JOURNALS OF GERONTOLOGY 51A(6):B403-B408 (1996)]. But if CRAN and IGF−1 reduction are a famine-like trigger of the same defensive metabolism in the organism — more heat shock protein, more antioxidant enzymes, greater DNA repair and reduced fertility (it would be wasteful to use resources on producing offspring who have little chance of surviving) — then additive effects of both would not be expected.
To say that IGF−1 and cell-signalling regulate aging is a half-truth — with the missing half required to explain the mechanisms of aging. What lies at the end of the cell signalling? The answer must be more antioxidant enzymes, more heat shock proteins and/or better DNA repair — and/or fewer harmful agents like cortisol and inflammatory cytokines. Aging is ultimately accumulated damage on the macromolecular, cellular and tissue level — ultimately the result of a limited number of possible defenses and a limited number of possible damaging agents. "Longevity genes" must result in decreased aging-damage or increased aging-damage repair. Although means may be found to enhance defenses to slow aging, the ultimate challenge is to find means to repair the damage.
(See Growth Hormone (GH/IGF−1) Replacement for more about GH and IGF−1.)
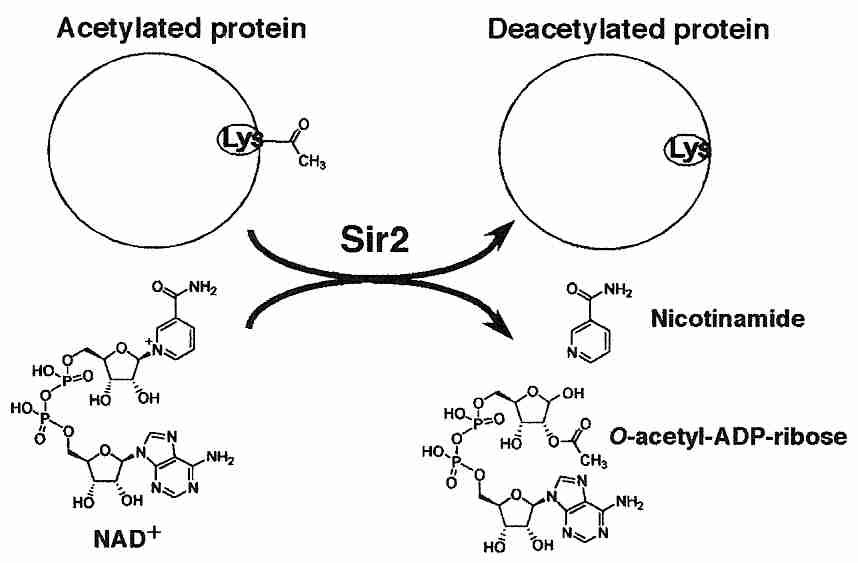
|
(For background on gene silencing by histone deacetylation, see Epigenetic Dysregulation.)
Like Drosophila and C. elegans, the budding yeast Saccharomyces cerevisiae (Brewer's yeast) has served as a model organism for aging research. Of interest has been the S. cerevisiae Silent Information Regulator (SIR2) gene which produces the Sir2 histone deacetylase protein. Acylation of histones reduces their binding to DNA, thereby facilitating transcription, whereas deacylation allows histones to bind to DNA thereby silencing gene expression. Replicative lifespan in yeast refers to the lifespan of a "mother" cell that buds-off "daughter" cells. The number of daughter cells that a mother produces before it "dies of old age" is called the replicative lifespan. The major cause of replicative aging is the "toxic" extra-chromasomal rDNA circles which segregate and accumulate in the mother cell. Yeast with SIR2 deletions have a short lifespan, whereas yeast with extra SIR2 genes have greatly extended lifespan [CELL; Longo,VD; 126(2):257-268 (2006)]. Gene silencing by deacetylation causes the chromatin to become more closed & inaccessible, thereby reducing genome instability [GENES & DEVELOPMENT; Guarente,L; 14(9):1021-1026 (2000)].
Yeast cells cease to divide under conditions of nutrient deprivation (severe calorie restriction). The survival of yeast under such conditions has been called chronological lifespan — and is regarded as a model for mammalian post-mitotic cells, in contrast to replicative lifespan, which is regarded as a model for mammalian proliferative cells. Deletion of SIR2 in yeast increases stress resistance and increases chronological lifespan when calories are restricted [PLoS GENETICS; Kaeberlein,M; 3(5):e84 (2007)].
SIR2 expression is activated by CRAN (Caloric Restriction with Adequate Nutrition), but neither CRAN nor extra SIR2 can silence genes without NAD+ (oxidized form of Nicotinamide Adenine Dinucleotide) as a co-factor [SCIENCE 289:2126-2128 (2000)]. The presence of a high NAD/NADH ratio in a cell is an index of low energy production. CRAN yeast with less NAD do not show greater longevity than ad-libitum fed yeast. The PNC1 gene encodes an enzyme which deaminates nicotinamide, and may be the upstream transducer of a variety of low-intensity, longevity-activating stressors, including heat, osmosis and CRAN — suggesting that life extension due to SIR2 is a generalized stress response [NATURE; Anderson,RM; 423:181-185 (2003)].
Insofar as genetic instability (ribosomal DNA recombination, in particular) seems to be the primary (fastest) "aging" mechanism in yeast, it is not surprising that gene silencing extends yeast lifespan. But ribosomal DNA is far more stable in higher organisms.
In C. elegans sir−2.1 — the gene most similar in DNA sequence to yeast SIR2 — inactivates the receptor for the nematode version of insulin — thereby activating Daf−16 protein production. Again, NAD+ is a necessary co-factor. Doubling the sir−2.1 gene in C. elegans apparently resulted in a 50% extension of lifespan [NATURE; Tissenbaum,HA; 410:227-230 (2001)].
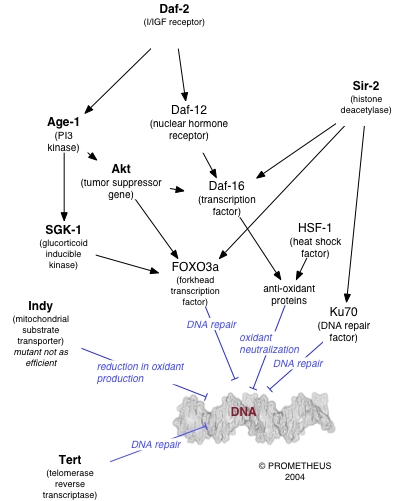
|
Experiments in which Drosophila Sir2 expression was quadrupled apparently led to a 57% extension of lifespan, with no further lifespan extension by CRAN. Flies on CRAN showed an increase in Sir2 mRNA and a lifespan increase that could be prevented by decreasing Sir2 gene function [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Rogina,B; 101(45):15998-16003 (2004)]. Drosophila fed the histone deacetylase inhibitor 4-phenylbutyrate showed up to 52% longer maximum lifespan. Gene analysis showed repressed expression of some metabolism genes and increased expression of genes for SOD, Elongation Factor−1-alpha (EF−1 α) & heat shock proteins, among others [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Kang,H; 99(2):838-843 (2002)].
In contrast to earlier research, however, a more careful analysis of Sir2 overexpression in C. elegans and Drosophila either showed no increase in lifespan [NATURE; Burnett,C; 477:482-485 (2011)], or a more modest 10-13% increase in lifespan [NATURE; Viswanathan,M; 477:E1-E2 (2011)].
Deacetylation of proteins (other than histones) can directly modify their activity. Most mammalian histone deacetylase enzymes are not NAD-dependent [JOURNAL OF PHARMACEUTICAL SCIENCES; Hisahara,S; 98(3):200-204 (2005)]. A family of NAD-dependent Sir2-like deacetylases in mammals called sirtuins includes SIRT1 in the nucleus, SIRT2 in the cytoplasm and SIRT3 in the mitochondria. The protein most resembling yeast SIR2 in humans is SIRT1. Unlike yeast Sir2 (which exclusively deacetlyates histones) SIRT1 has a wide range of substrates [GENES & DEVELOPMENT; Haigis,MC; 20(21):2912-2921 (2006)] including p53 and FOXO3 protein which SIRT1 silences by direct deacylation — reputed to prevent "premature senescence" and "premature apoptosis". Nonetheless, SIRT1 deacetylation of p53 has not been shown to alter cell survival following DNA damage [MOLECULAR AND CELLULAR BIOLOGY; Solomon,JM; 26(1):28-38 (2006)].
Polyphenols such as quercetin, resveratrol (especially) and other sirtuin-activating compounds have been reported to extend the lifespan of nematodes & fruit flies. Resveratrol was shown to extend lifespan of nematodes by 10% and fruit flies by 29% without loss of fertility [NATURE; Wood,JG; 430:686-689 (2004)]. No lifespan increase was seen in flies lacking functional Sir2 and no additional life extension was seen with CRAN. This led to the conclusion that (unlike C. elegans daf−2 mutations), the life-extending benefits of Sir2 operate by a similar mechanism as CRAN. But there is experimental evidence contradicting this conclusion [SCIENCE; Kaeberlein,M; 312:1312 (2006)].
Resveratrol has also been shown to extend the lifespan of short-lived fish (killifish) nearly 60% without loss of fertility [CURRENT BIOLOGY; Valenzano,OR; 16(3):296-300 (2006)], possibly by increased sirtuin expression, but a number of other mechanisms are possible [MUTATION RESEARCH; Kundu,JK; 555(1-2):65-80 (2004) and JOURNAL OF BIOLOGICAL CHEMISTRY; Kaeberlain,M; 280(17):17038-17045 (2005)].
Analysis of the brain, kidney, liver and other tissues of rats subjected to 60% ad libitum CRAN showed increased SIRT1 expression [SCIENCE; Cohen,HY; 305:390-392 (2004)]. Treatment of human embryonic kidney cells with resveratrol or transfection of those cells with SIRT1 expression vector resulted in a dose-dependent reduction of Bax protein mediated apoptosis [SCIENCE; Cohen,HY; 305:390-392 (2004)]. SIRT1 increases FOXO3's induction of cell cycle arrest and resistance to oxidative stress, but inhibits FOXO3's induction of apoptosis [SCIENCE; Brunet,A; 303:2011-2015 (2004)].
Methods of increasing NAD or activating sirtuins have been proposed to prevent neurodegeneration [SCIENCE; Araki,T; 305(5686):1010-1013 (2004)]. Neurons are post-mitotic (as are all cells in fruit flies and nematodes) so the anti-apoptotic effect of SIRT1 would have to be the main pro-survival mechanism. In response to DNA damage, SIRT1 may inhibit p53-mediated cell-cycle arrest and apoptosis, but in response to TNF−α cytokine reduces NF−κB mediated inhibition of cell-cycle arrest and apoptosis [THE EMBO JOURNAL; Yeung,F; 23(12):2369-2380 (2004)]. Resveratrol similarly reduces NF−κB expression.
Ku70 protein, which functions in DNA repair, is normally bound tightly to Bax protein in the cytoplasm, but in response to stress Ku70 is activated and releases Bax, which can then move into the mitochondria to initiate apoptosis. SIRT1 reduces ku70 acetylation and thereby opposes apoptosis [SCIENCE; Cohen,HY; 305:390-392 (2004)]. If SIRT1 were increasing lifespan by resisting apoptosis, the effect would be similar to that seen in p66shc mice. Human SIRT6 maintains telomere integrity [NATURE; Michishita,E; 452:492-496 (2008)].
Sirtuins facilitate NHEJ (Non-Homologous End-Joining DNA repair mechanisms for Double-Strand Breaks, DSBs) by an unknown mechanism [ACTA BIOCHIMICA POLONICA; Wojewodzka,M; 54(1):63-69 (2007)]. ATM phosphorylation of H2AX recruits SIRT1 to DSBs, which evidently assists in repair [CELL; Oberdoerffer,P; 135(5):907-918 (2008)]. Derepression of the genome at loci vacated by SIRT1 recruited by genotoxic stress may lead to generalized dysdifferentiation associated with aging. In vitro study of gene expression in neocortical tissue showed that more than two-thirds of SIRT1-bound genes derepressed during aging were also derepressed by oxidative stress [CELL; Oberdoerffer,P; 135(5):907-918 (2008)].
SIRT1 also represses the nuclear receptor PPAR−γ (Peroxisome Proliferator-Activated Receptor−gamma), thereby triggering lipolysis and loss of fat, suggestive of the life-extending benefits of fat reduction in FIRKO mice. Treatment of fibroblasts with the SIRT1 activator resveratrol resulted in a significant reduction of fat content [NATURE; Picard,F; 429:771-776 (2004)]. PPAR−γ inhibition has been used to block the development of insulin resistance due to obesity and type 2 diabetes [AMERICIAN JOURNAL OF PHYSIOLOGY; Miles,PDG; 284(3):E618-E626 (2003)], suggestive of the idea that CRAN and the metabolic syndrome are opposite ends of a continuum.
A "neurohormonal clock" in the brain of mammals has been suggested to influence aging through neurohormones. Hormones alter the gene expression of DNA throughout the body. The pituitary gland (the "master gland") under the influence of the brain/hypothalamus can thus influence the physiology of all body cells. When the pituitary gland of mammals is surgically removed and supplements of essential hormones are given, maximum lifespan increases by one third to one half. Such mammals voluntarily reduce their caloric intake, which suggests that the life extension may be primarily due to CRAN (Caloric Restriction with Adequate Nutrition).
The hormones DHEA, melatonin, thyroid, and somatotropin (Growth Hormone, GH) decline with age. Women experience menopause, with the loss of progesterone and estradiol secretion from the ovaries. Specific areas of the brain show age-related declines in the levels of the neurotransmitters dopamine, acetylcholine, norepinephrine, GABA and serotonin. With aging there is a decline in both serotonin transporters [LIFE SCIENCES; Yamamoto,M; 71(7):751-757 (2002)] and serotonin receptors [NEUROPSYCHOPHARMACOLOGY; Meltzer,MD; 71(7):751-757 (2002)]. Serotonin is the precursor for melatonin in the brain.
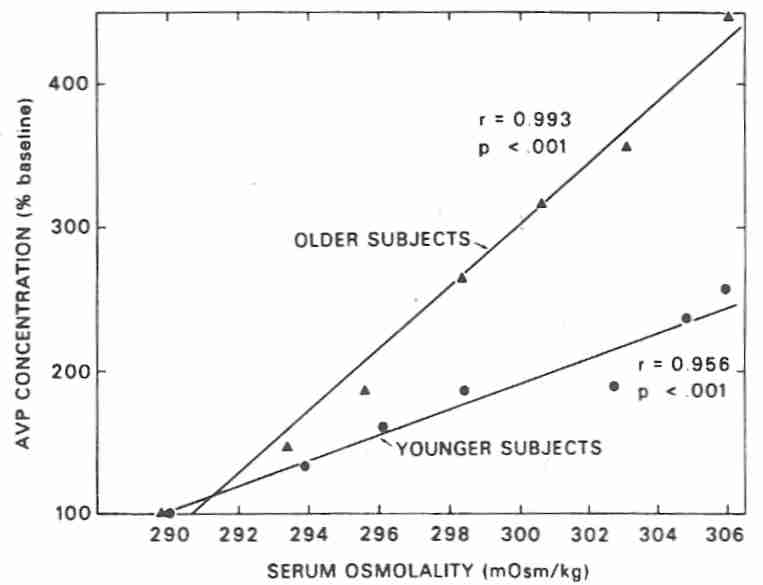
|
Aging is associated with increasing secretion of the hormone 8−arginine vasopressin (AVP, also known as Anti-Diuretic Hormone or simply vasopressin), and a decreasing ability of AVP to increase serum osmolality. With age there is a decline in kidney AVP receptors, which results in increased AVP secretion and decreased AVP effectiveness [AMERICAN JOURNAL OF PHYSIOLOGY; Tian,Y; 287(4):F797-F805 (2004)]. Nearly 10% of the elderly suffer from hyponatremia, and nearly twice as many elderly nursing home residents suffer from that affliction [JOURNAL OF THE AMERICAN GERIATRIC SOCIETY; Miller,M; 54(2):345-353 (2006)].
According to the glucocorticoid cascade hypothesis, glucocorticoid steroid hormones show rising blood levels with age, which increasingly damages feedback inhibition neurons in the hippocampus, resulting in even greater increases of blood glucocorticoid and a destructive feedback loop. Glucocorticoid hormone (cortisol in humans) is a normal response to stress. Cortisol mobilizes blood glucose and depresses the immune/inflammatory response, among other effects. Although useful in emergencies, chronic stress can be catabolic (destructive — Pacific salmon use glucocorticoids to self-destruct after spawning). Physical & psychological stress causes the brain to release Corticotropin Releasing Factor (CRF) & vasopressin — both of which stimulate pituitary release of AdrenoCorticoTropic Hormone (ACTH). ACTH causes the adrenal cortex to release glucocorticoids.
High blood levels of glucocorticoids are sensed by neurons in the hippocampus, which signal the brain to release less vasopressin. The involvement of hippocampal neurons makes sense because stressful situations are often associated with vivid & detailed memories. Patients with major depression can lose 20% of their hippocampal volume. Cortisol can reduce neuron uptake of glucose by 15-25% — which can contribute to neuron death [EXPERIMENTAL GERONTOLOGY 34:721-732 (1999)]. Moreover, glucocorticoids reduce cellular SOD & glutathione peroxidase activity in all brain areas [BRAIN RESEARCH 791:209-214 (1998)].
Although the glucocorticoid cascade hypothesis is not a theory of aging in the sense of reducing lifespan, it does count as a theory of brain aging — declining capacity for memory-formation in particular. Blood glucocorticoid increases with age in rats, but humans normally do not show increasing levels of glucocorticoid until the late 70s or 80s. About half of Alzheimer's Disease patients show significantly elevated cortisol, however. Most people do normally show increasing number (and perhaps hypertrophy) of neurons in the ParaVentricular Nucleus (PVN) — which expresses both CRF & vasopressin — as they age. Evidence suggests an inverted U−shaped relationship between cortisol & cognition — and that sustained higher cortisol levels can lead to a non-Alzheimer's dementia in humans [JOURNAL OF PSYCHIATRIC RESEARCH 35:127-145 (2001)]. Estrogen can prevent or even reverse cortisol-induced brain damage [BRITISH JOURNAL OF CLINICAL PHARMACOLOGY 52:647-653 (2001)].
Much antioxidant protection in the brain is due to bilirubin which is produced by Heme Oxygenase (HO) enzyme, particularly in the hippocampal neurons. HO expression declines with age in the rat brain, and this decline has been linked to elevated glucocorticoid expression [JOURNAL OF NEURAL TRANSMISSION; Ewing,JF; 113(4):439-454 (2006)]. Additionally, HO has antiapoptotic effects which are independent of the antioxidant effects of bilirubin [KIDNEY INTERNATIONAL; Nath,KA; 70(3):432-443 (2006)]. (For more on the antioxidant effects of bilirubin see Antioxidant Enzymes.)
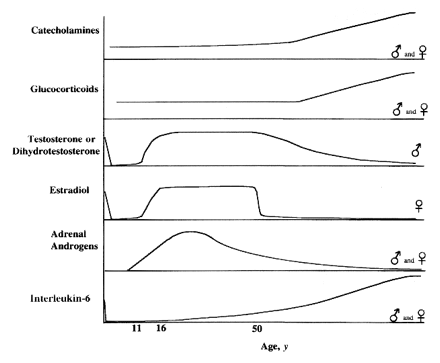
|
Some critics have pointed-out that CRAN animals show elevated glucocorticoids, but it should come as no surprise that calorie-restriction is stressful. In fact, the glucocorticoid cascade hypothesis raises the worrisome possibility that CRAN could prolong lifespan while simultaneously undermining memory capabilities. But experiments on CRAN animals have not shown learning deficiencies, quite the opposite.
Humans and other primates are the only species that produce & secrete the hormone DeHydroEpiAndrosterone (DHEA) and it's sulfate (DHEA−S) in quantities surpassing those of any other steroid. DHEA levels peak in the late 20s and decline to 10% of the peak by age 80. DHEA may protect against the harmful effects of cortisol while contributing to androgen & estrogen synthesis in peripheral tissues, promoting lean body mass, reducing depression and improving immune function [EXPERIMENTAL GERONTOLOGY 33(7/8):713&897 (1988)].
Growth Hormone (GH) also declines with age (about 14% per decade after age 25), which is blamed for increased fat deposition, loss of muscle mass and bone demineralization. There is evidence that GH replacement can improve cardiovacular health, boost immune function and improve cognitive function in older adults, but there is also the danger that GH replacement can increase insulin resistance and cancer risk.
The idea of restoring all hormones and growth factors to youthful levels as a means of rejuvenation has a strong intuitive appeal, but hormones often have the risk of promoting cancer growth. Given the fact that cancer incidence increases with age, declining hormone levels may even contribute to elderly survival. Only when cancer is eliminated will replacement of all age-declining hormones be safe. Even then, however, for cases where declining receptor sensitivity rather that declining hormone release are associated with age (as with AVP), hormone replacement will not get to the root of the problem — and can be harmful without causing cancer.
[For more about DHEA and GH, see DHEA Hormone Replacement and Growth Hormone (GH/IGF−1) Replacement].
[For more about sex hormone replacement, see Sex Hormone Replacement in Older Adults]
According to the "immune system theory of aging", many aging effects are due to the declining ability of the immune system to differentiate "foreign" from "self" proteins. Not only does the immune system become less capable of resisting infection & cancer, but declining cell function could be due to attacks by the immune system against native tissues. Arthritis, psoriasis and other autoimmune diseases increase with age. There is evidence that histocompatability genes, genes affecting DNA repair and genes for SOD production — all of which affect longevity — are located close together on human chromosome 6.
Leukocytes (white blood cells), which form the basis of the immune system (along with complement proteins), are roughly 65% granulocytes (mostly neutrophils), 5% monocytes (which can become macrophages) and 30% lymphocytes. Lymphocytes can be subclassified as B−lymphocytes (B−cells) or T−lymphocytes (T−cells) based on whether they mature in Bone marrow or the Thymus gland (all lymphocytes originate in bone marrow). Antigens are molecular portions of pathogens that act as identifiers. B−cells generate antibodies ("humoral immunity") against antigens, whereas T−cells directly bind to antigens ("cellular immunity").
The thymus gland of the immune system reaches its greatest weight during puberty, and shrinks thereafter, with lymphoid tissue being replaced by fat. The shrinking of the thymus gland proceeds far more rapidly than the progress of aging — at age 50 the thymus of humans is typically only 5−10% of its original mass. Nonetheless, T−cells remain fairly constant over most of adult life due to peripheral proliferation (although proliferation declines in the elderly).
Because the thymus is the organ in which T−cells "matures", once maturation occurs most of the work of the thymus is done. In the maturing T−lymphocyte system, the thymus creates a broad diversity of T−cells, each of which is programmed to recognize and combat a different antigen. T−cells which would combat self-substances are eliminated by apoptosis.
The immune system uses proliferation & apoptosis to create & refine T−cells. The immune system uses clonal expansion (rapid multiplication of lymphocytes of a single "clone" against a single antigen) & apoptosis to control the numbers of T−cells available to fight specific antigen threats. Injecting the protein Apo−1 into a cell will trigger apoptosis. But the protein Bcl−2 can rescue a cell from apoptosis. With aging, mature T−cells increasingly manifest apoptosis for reasons that seem to be unrelated to decreased Bcl−2 expression or oxidative stress [MECHANISMS OF AGING AND DEVELOPMENT; Phelouzat,M; 88:25-38 (1996)].
T−lymphocytes that have not encountered an antigen since creation are called naive T−cells, whereas T−lymphocytes that have been clonally expanded to fight an invading antigen are called memory T−cells. T−cells of the elderly have a much higher ratio of memory T−cells to naive T−cells than younger people. Old memory T−cells have less CD28 surface protein than young memory T−cells and are thus less able to divide when presented with antigen [JOURNAL OF IMMUNOLOGY; Engwerda,CR; 152:3740-3747 (1994)]. CD28 ligation is required for production of IL−2 cytokines. The elderly memory T−cells have short telomeres and are thought to accumulate because of increasingly defective apoptosis [IMMUNOLOGIC RESEARCH 21(1):31-38 (2000)].
Two predominant forms of T−cells are cytotoxic T−cells (with CD8 surface receptors) and helper T−cells (with CD4 surface receptors). The cytotoxic T−cells attack bacteria or cancerous cells by punching holes in the cells and injecting them with toxic proteins. The helper T−cells secrete growth factors (cytokines) that foster the clonal expansion of other T−cells and/or of antibody-producing cells (the B−lymphocytes). Helper T−cells are more numerous in youth & maturity, but in the elderly the ratio of CD8 to CD4 cells increases. CD8 T−cells become more resistant to apoptosis with aging, whereas CD4 cells become more susceptible to apoptosis.
Cytomegalovirus prevalence significantly increases in the elderly, and may be responsible for much of the skewed CD8:CD4 ratio of advanced age [JOURNAL OF IMMUNOLOGY; Hadrup,SR; 176(4):2645 (2006)]. Cytomegalovirus infection increases with age in humans [CLINICAL INFECTIOUS DISEASES; Staras,SAS; 43(9):1143-1151 (2006)], primarily infects antigen-presenting cells & can increase inflammatory cytokines [REVIEWS IN MEDICAL VIROLOGY; Varani,S; 19(3):131-145 (2009)], and is associated with immunosenescence [CURRENT OPINION IN IMMUNOLOGY; Derhovanessian,E; 21(4):440-445 (2009)]. Naive CD4 cells decline rapidly after age 65 [JOURNAL OF IMMUNOLOGY; Naylor,K; 174(11):7446-7452 (2005)].
There are two types of helper T−cells, designated TH1 (type 1) and TH2 (type 2). The TH1 cells promote growth of T−lymphocytes with the cytokine InterLeukin−2 (IL−2), whereas the TH2 cells promote growth of B−lympocytes with the cytokine InterLeukin−4 (IL−4). TH1 cells are more prominent in autoimmune infections, whereas TH2 cells are more prominent in viral infections. In youth & maturity the TH1 cells predominate, but in the elderly the TH2 cells predominate [MECHANISMS OF AGING AND DEVELOPMENT 94:1-5 (1997)]. Moreover, aging is accompanied by a significant loss of IL−2 as well as of IL−2 receptors — a phenomenon thought to be responsible for the significant decline of proliferation (clonal expansion) in response to antigens seen with aging [SCIENCE 273:70-74 (1996)]. The decline of T−cell activation due to reduced IL−2 production is at least partially due to oxidation-damaged proteasomes being less capable of inducing the gene transcription factor NFκB [CELLULAR IMMUNOLOGY 192:167-174 (1999)].
Proliferation of T−cells in response to antigenic or mitogenic (cell-division stimulating) signals also declines with aging — apparently due to to decline in activity of the Mitogen Activating Protein Kinase (MAPK) cascade which causes cell surface signals to alter gene expression. CRAN (Caloric Restriction with Adequate Nutrition) significantly reduces the decline of MAPK activity associated with aging [PROCEEDINGS OF THE SOCIETY FOR EXPERIMENTAL BIOLOGY AND MEDICINE 223:163-169 (2000)]. But selenium supplementation has been shown to restore lymphocyte proliferation in aged mice to that of normal young adults [PROCEEDINGS OF THE SOCIETY FOR EXPERIMENTAL BIOLOGY AND MEDICINE; Roy,M; 209(4):369-375 (1995)].
The combination of low T−cell proliferation and low CD4/CD8 ratio was highly predictive of low 2−year survival in a study of people in the 86−92 age range [JOURNALS OF GERONTOLOGY 50A(6):B378-B382 (1995)]. Melatonin elevates the CD4/CD8 ratio [IMMUNITY & AGEING; Srinivasan,V; 2:17 (2005)]. Immune function is very important for the elderly because infection causes an increasing percentage of deaths for those over 80 years of age [JOURNALS OF GERONTOLOGY 52A(1):B67-B77 (1997)] and AMERICAN JOURNAL OF MEDICINE; 114:365-369 (2003)].
Regulatory T−cells that suppress T−cell-mediated autoimmune diseases in humans decline with age [JOURNAL OF NEUROSCIENCE RESEARCH; Tsaknaridis,L; 74(2):296-308 (2003)].
Natural Killer (NK) cells differ from cytotoxic T−cells by the ability to lyze pathogenic cells without the need of antigens. NK cells decline in activity with age, but this decline is compensated for by an increase in NK cell numbers. In centenarians, however, no decline in NK activity has been seen — nor was there a decline in youthful CD8/CD4 cells [IMMUNOLOGY TODAY; Franceschi,C; 16(1):12-16 (1995)].
B−cells from older animals produce less antibody and express less of the surface CD40 protein which causes B−cell activation and differentiation. The decline in T−cell activity with age is responsible for most of the decline in B−cell numbers and activity.
Macrophages are immune-system cells that "eat" foreign particles (including bacteria) and digest the particles in lysosomes. Monocytes are the small blood stream cells that swell to become macrophages after migrating into tissues. Monocytes from elderly humans have a greatly reduced capacity to produce the cytokine InterLeukin−1 (IL−1) and the toxic free radicals that macrophages use to kill foreign or cancerous cells [THE JOURNAL OF IMMUNOLOGY 154:832-843 (1995)]. Nonetheless, the superoxide, hydrogen peroxide, hydroxyl ions & nitric oxide produced by neutrophils & macrophages to kill bacteria can attack native tissues in age-associated chronic inflammation. The reactive products of nitric oxide and oxygen species inhibit PARP-mediated DNA repair [FREE RADICAL BIOLOGY & MEDICINE 35(11):1431-1438 (2003)].
Some of the decline in immune function in the elderly may be due to protein cross-linking in tissues & blood vessels reducing immune-cell mobility and access to infected areas. Poor nutrition in the elderly is also a factor. Supplements consisting of recommended dietary allowances of nutrients (plus extra Vitamin E & beta-carotene) significantly improved the immune status of elderly subjects [THE LANCET 340:1124-1127 (1992)]. Supplementation with the steroid hormone DeHydroEpiAndrosterone (DHEA, a hormone that dramatically declines with age) increased IL−2 & Interferon-gamma activity in mice [THE JOURNAL OF INFECTIOUS DISEASES 167:830-840 (1993)].
Vulnerability to death by influenza & pneumonia increases rapidly with age in the United States. A person aged 50−64 is nearly ten times more likely to die from an influenza-associated death as a person in the 5−49 age group. And a person over 65 is over ten times more likely to die from an influenza-associated death as a person in the 50−64 age group. A person over 85 is about 16 times more likely to die an influenza-associated death as a person in the 65−69 age group [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION; Thompson,WW; 289(2):179-186 (2003)]. Vaccination of the elderly reduces influenza-associated death by 50% [ARCHIVES OF INTERNAL MEDICINE; Hak,E; 165(3):274-280 (2005)].
With aging the body contains increasing quantities of proinflammatory cytokines such as TNF−α, IL−1 and IL−6, which is positively associated with cardiovascular disease mortality [IMMUNOLOGY AND ALLERGY CLINICS OF NORTH AMERICA; Bruunsgaard,H; 23(1):15-39 (2003)]. The increase in memory cells results in an increase in the cytokines IL−4 & IL−10 that are produced by the memory cells. Lifetime exposure to infectious disease reduces lifespan by accelerated immunosenescence [FEBS LETTERS; Martinis,MD; 579:2035-2039 (2005)] and chronic inflammation [SCIENCE; Finch,CE; 305:1736-1739 (2004)]. Chronic inflammation is implicated in atherosclerosis, arthritis, Alzheimer's Disease, cancer, the metabolic syndrome (type 2 diabetes) and numerous other afflictions affecting the elderly. Inflammation is probably not the major cause of the damage & degeneration of aging, but it contributes to the damage. Free radicals and oxidized glycation products (AGEs) are contributers to chronic inflammation.

|
Aging is associated with increasing activity of the pro-inflammatory transcription factor NF-κB (NF−κB). NF−κB is normally bound to IκB protein in the cytoplasm, but is released to enter the nucleus when infection, oxidative stress or pro-inflammatory cytokines cause ubiquitination and subsequent protease degradation of IκB. NF−κB increases transcription of genes coding for TNF−α and IL−1, which can result in a positive feedback loop. The ability of free radicals (ROS, Reactive Oxygen Species) to cause NF−κB release and the production of ROS by inflammation also results in a positive feedback loop. NF−κB and TNF−α are central to the aging-associated increase in chronic inflammation. Although glucocorticoids are increased in aging & CRAN and can inhibit NF−κB, stimulation of NF−κB by stressors predominates. Not only does NF−κB release increase with age, but aging results in NF−κB binding more strongly to DNA [BIOCHEMICAL JOURNAL; Helenius,M; 318(Pt 2):603-608 (1996)]. Age-associated increases in ceramide results in increased NF−κB activation [THE JOURNAL OF IMMUNOLOGY; Wu,D; 179(7):4829-4839 (2007)].
NF−κB induced chronic inflammation in combination with its ability to suppress apoptosis (inhibiting the elimination of cancer cells) often leads to cancer [NATURE IMMUNOLOGY; Karin,M; 3(3):221-227 (2002)]. Cancer can also be initiated by NF−κB induction of inducible Nitric Oxide Synthetase (iNOS), leading to DNA damage, and the inhibition of apoptosis by NF−κB again favors cancer [NATURE REVIEWS, IMMUNOLOGY; Karin,M; 5:749-759 (2005)].
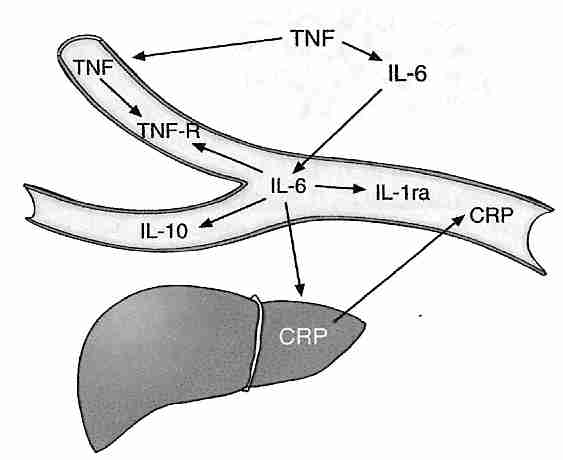
|
Aside from the induction of TNF−α (Tumor Necrosis Factor-alpha) by NF−κB, TNF−α is produced by visceral fat. Obese people can produce twice as much TNF−α as lean people produce. White adipose tissue attracts macrophages, which produces the inflammatory agents (like TNF−α) associated with obesity-induced insulin resistance [JOURNAL OF CLINICAL INVESTIGATION; Xu,H; 112(12):1821-1830 (2003)]. Surgical removal of visceral fat (but not subcutaneous fat) extends the mean and maximum lifespan of rats [ BIOCHEMICA ET BIOPHYSICA ACTA; Huffman,DM; 1790(10):1117-1123 (2009)]. TNF−α can induce apoptosis, but only if protein synthesis is inhibited [SCIENCE; Beg,AA; 274:782-784 (1996)].
TNF−α upregulates NF−κB and IL−6 (InterLeukin−6). IL−6 upregulates pro-inflammatory cytokine IL−1 and induces the liver to produce the inflammatory protein CRP (C−Reactive Protein). Proinflammatory cytokines have been shown to induce cellular senescence [FREE RADICAL RESEARCH; Sasaki,M; 42(7):625-632 (2008) and CELL; Kuilman,T; 133(6):1019-1031 (2008)]. But IL−6 but also induces production of the anti-inflammatory cytokine IL−10 while inhibiting TNF−α. IL−10 (which inhibits TNF−α production) is produced in larger quantities when exogenous S-adenosylmethionine is administered [AMERICAN JOURNAL OF PHYSIOLOGY; Song,Z; 284(6):G949-G955 (2003)]. CRP is an important risk factor for myocardial infarction (heart attack). A four-year study of women showed those in the highest quarter of blood CRP had 15.7 times greater risk of developing type 2 diabetes as those in the lowest quarter [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION; Pradham,AD; 286(3):327-334 (2001)]. In a study of men, those in the highest quarter of blood CRP had 3 times the risk of developing dementia as those in the lowest quarter [ANNALS OF NEUROLOGY; Schmidt,R; 52(2):168-174 (2002)].
Increasing plasma levels of pro-inflammatory cytokines with aging can induce a stress response that is responsible for the increased plasma levels of cortisol associated with aging [INFLAMMATION RESEARCH; Sergio,G; 57(12):558-563 (2008)]. Although elevated cortisol is generally anti-inflammatory in the periphery, cortisol can be pro-inflammatory in the hippocampus and cerebral cortex [BRAIN, BEHAVIOR, AND IMMUNITY; Sorrells,SF; 21(3):259-292 (2007)].
Exercise can be very anti-inflammatory by increasing muscle-derived IL−6 production (which is independent of TNF−α) and reducing CRP [JOURNAL OF APPLIED PHYSIOLOGY; Peterson,AWW; 98(4):1154-1162 (2005)]. Adequate sleep can reduce TNF−α and IL−6 secretion (both of which induce sleepiness & fatigue). Reduction of IL−6 production by the administration of sex steroids has been suggested as a means of reducing problems with sleepiness & fatigue in the elderly [THE JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM; Vgontias,AN; 88(5):2087-2095 (2003)]. DHEA can also reduce IL−6 production [THE JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM; Straub,RH;83(6):2012-2017 (1998)].
CycloOXygenase (COX) enzyme activity increases with age, thereby increasing the production of prostaglandins that inhibit T−cell proliferation. Increased levels of hydrogen peroxide probably are responsible for the age-related increase in COX activity, indicated by the fact that Vitamin E attenuates COX activity and restores T−cell proliferation [AMERICIAN JOURNAL OF PHYSIOLOGY; Wu,D; 275(3 Pt 1):C661-C668 (1998)].
Advanced Glycation End-products (AGES) not only originate from metabolism, but can be ingested in diet or tobacco smoke and contribute significantly to inflammation. AGEs can activate NF−κB [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Vlassara,H; 99(24):15596-15601 (2002)]. High blood insulin potentiates NF−κB in a dose-dependent manner [CIRCULATION RESEARCH; Golovchenko,I;87(9):746-752 (2000)]. Increased NF−κB activity by AGEs is often mediated by a Receptor for AGE (RAGE), which can also be activated by TNF−α [JOURNAL OF BIOLOGICAL CHEMISTRY; Tanaka,N; 275(33):25781-25790 (2000)]. NF−κB activated by oxidative stress or AGEs upregulates the expression of RAGE (more AGE receptors), creating a positive feedback loop that worsens chronic inflammation [CIRCULATION RESEARCH; Schmidt,AM; 84(5):489-497 (1999)].
Even though age-associated chronic diseases are important components of segmental progerias , they are often dissociated from aging per se because those people who achieve maximum lifespan typically do not die of these diseases. The role of controllable risk factors like obesity, exercise, AGE ingestion and Vitamin E supplementation would also tend to dissociate inflammation and chronic disease from a central role in the essential aging process. Nonetheless, these chronics diseases are aging-associated and it is likely that inflammation plays some role in the degenerative & damaging processes known as aging.
Many chemicals accumulate in the cells with age, including toxic & inert substances from the exterior and similar substances arising as byproducts of cellular metabolism [notably Advanced Glycation End-products (AGEs) and lipid peroxidation debris]. Fat-soluble substances (such as DDT & PCBs) are particularly slow to be eliminated. Iron tends to accumulate in cell nuclei with aging, as does aluminum. Aluminum transforms metabolically active DNA into an inert state. Lead also accumulates in cells, and is neurotoxic. Cytochrome P−450 detoxification enzymes of the liver (which have maximal light absorption at 450 nanometer wavelength) decline with age. In the 1976 to 1980 period, the 15% of US population with the highest blood levels of lead had 49% higher cardiovascular mortality and 68% higher cancer mortality [ARCHIVES OF INTERNAL MEDICINE; Lustberg,M; 162(21):2443-2449 (2002)].
Non-dividing cells (muscle cells, heart muscle cells and neurons) are not susceptible to the Hayflick Limit. Nor is double-chromosome damage of as great concern in non-dividing cells as it is for dividing cells. But for non-dividing cells that cannot be replaced — heart muscle cells and neurons — the accumulation of cellular garbage may be a very significant factor in cellular aging. Species survival may be thus dependent on the creation of new organisms once the old ones have accumulated too much chemical garbage to be functional.
Of particular note is lipofuscin (age pigment), which can accumulate in large quantities in non-dividing cells. Lipofuscin is regarded as a product of lysosomes — organelles containing hydrolytic enzymes to degrade proteins, lipids and damaged organelles. As production of lysosome enzymes decline with age — and as lysosomes engulf increasingly cross-linked proteins & lipids that are resistant to enzyme degradation — dysfunctional lysosomes (bloated with indigestible contents) accumulate in cells containing lipofuscin granules. Lipofuscin granules (engorged lysosomes) are characterized by a single membrane envelope, enclosing yellowish-brown material that can autofluorescence.
Inhibitors of proteases (enzymes that degrade protein) and Vitamin E deficiency result in lipofuscin-like cellular residues — a clue to the origin of lipofuscin. There is evidence that lipofuscin formation inhibits protein degradation, thereby creating a vicious cycle that promotes its own formation [EXPERIMENTAL GERONTOLOGY 36:475-486 (2001)]. In contrast to ceroids — which rapidly accumulate extra− & intra− cellularly in pathologic conditions — lipofuscin accumulates slowly, universally and specifically accumulates in lysosomes [ANNALS OF THE NEW YORK ACADEMY OF SCIENCES; Portas,EA; 959:57-65 (2002)]. The composition of lipofuscin — nearly half protein, one-third carbohydrate and the rest lipid — indicates that it is primarily composed of AGEs rather than lipid peroxidation products [BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS 236:327-332 (1997)].
Lipofuscin is normally diluted-out of dividing cells, although it is seen in increasing amount in fibroblasts nearing the Hayflick Limit. Lipofuscin accumulation in the non-dividing cells of the brain&heart is very prominent and is, in fact, regarded as a biomarker of aging. Lipofuscin accumulation in retinal pigment epithelial cells may lead to age-related macular degeneration, the leading cause of blindness in the developed world. The fact that lipofuscin accumulates at a higher than normal rate in Alzheimer's Disease and the fact that the disease is also characterized by abnormal tau-protein and amyloid-protein suggests that creation of defective protein and/or problems with removal of defective protein could be the underlying cause of Alzheimer's Disease.
Aging due to free-radicals & glycation of macromolecules other than DNA would be expected more in non-dividing cells than dividing cells — most notably in neurons. That lipofuscin is a component of neuron aging due to free-radical damage is indicated by the high levels of metals (especially iron) in lipofuscin. Oxidative stress has been shown to promote lipofuscin formation, whereas antioxidants reduce lipofuscin formation [FREE RADICAL BIOLOGY & MEDICINE 33(5):611-619 (2002)]. Although antioxidants cannot extend maximum lifespan of organisms as a whole, they may extend the maximum lifespan of neurons or even the entire brain. If so, antioxidants combined with organ replacement could be a means of extending maximum lifespan.
Lysosomes are normally responsible for degradation of aging mitochondria. But as lysosomes become increasingly dysfunctional due to accumulation of indigestible lipofuscin, cells become increasingly populated with aging, swollen mitochondria that produce less energy and more superoxide. Reactive oxygen species produce more aldehydes and more aldehyde-bridges between proteins, resulting in more lipofuscin [EUROPEAN JOURNAL OF BIOCHEMISTRY 269(8):1996-2002 (2002)]. There is thus a positive feedback loop of lipofuscin production, impaired lysosomes, dysfunctional mitochondria and aldehyde formation.
As a cause of death the relative incidence of cancer increases exponentially to age 65 and decreases thereafter. At age 65, 30% of North American deaths are due to cancer, whereas at age 80 only 12% of deaths are due to cancer — mostly because the relative increase of cardiovascular and Alzheimer's Disease is faster than the increase in cancer with age. Nonetheless aging is a major risk factor for cancer, and aging is associated with cancer.
But aging can also be distinguished from cancer, much as with other diseases associated with aging such as atherosclerosis, Alzheimer's Disease, osteoporosis and arthritis. In children, cancers are predominantly leukemias, lymphomas and sarcomas, whereas 80% of adult cancers in the United States are carcinomas. Nearly 90% of mice die of cancer, with about 2/5 of those cancers being lymphomas in males and about 3/5 lymphomas in females. About 30% of male mouse cancers are carcinomas and about 40% are sarcomas [RADIATION RESEARCH; Tanaka,IB; 167(4):417-437 (2007)]. In Werner's Syndrome sarcomas (connective tissue malignancies, usually) are more common than carcinomas. As with the mouse, this may be due to cellular immortalization by ALT rather than telomerase. These patterns do not indicate a simple relationship between aging and cancer.
That there is a distinction between aging and cancer is suggested by the fact that ionizing radiation increases cancer rate, but has less (if any) effect on the rate of aging. Atomic bomb survivors [RADIATION RESEARCH; Preston,DL; 160(4):381-407 (2003) and populations living near a nuclear test site [RADIATION RESEARCH; Bauer,S; 164(4 Pt 1):409-419 (2005)] showed increased noncancer mortality from aging-associated diseases (stroke, heart disease, respiratory disease), but there is no proof that this constituted accelerated aging. Experimental animals subjected to chronic sublethal ionizing radiation (alpha−, beta−, gamma− & X−rays that cause atoms & molecules to form ions) have shown generalize atrophy ("premature aging") and shortened lifespans, but single X−ray & ionizing radiation exposures have more noticeably increased kidney degeneration and cancer (especially leukemia). Other mutagens increase the risk of tumor-formation without reducing maximum lifespan. These results indicate that spontaneous mutations & chromosome breakage are not normal contributors to aging. Mutations due to ionizing radiation are qualitatively different from those occurring "spontaneously" with the passage of time [MUTATION RESEARCH 375:37-52 (1997)]. Mammals with longer lifespans have been shown to have more efficient DNA repair of gamma-radiation [EXPERIMENTAL GERONTOLOGY 37:1203-1205 (2002)]. Nonetheless, the view that ionizing radiation causes accelerated aging is not easily dismissed [AGING; Richardson,RB; 1(11):887-902 (2009)].
Cancer is a disease of DNA, whereas aging is a disease of all organs, tissues, cells and macromolecules. Most cancers are caused by chemical carcinogens, which may result in DNA damage different from DNA damage associated with aging. Cancer is a disease of dividing cells — especially the rapidly dividing cells of the epithelium & blood-forming tissues. Non-dividing cells like neurons or muscle cells don't become cancerous, but aging affects all tissues. A study of 15 rodent species showed that telomerase repression is a feature of large size rather than long life, suggesting that tumor initiation usually occurs during growth and development [AGING CELL; Seluanov,A; 6(1):45-52 (2007)]. Telomerase repression rather than replicative senescence can be the primary anti-cancer mechanism.
DNA must ultimately be responsible for the great variation of maximum lifespan between species. But in this respect DNA (the genome) partly is responsible for the production of reactive oxygen species as well as for the capacity of tissues to withstand oxidative stress & glycation as well as other chemical challenges. If aging is distinguished from cancer by toxin/garbage accumulation and by damage to all macromolecules rather than just DNA, it is nonetheless true that the DNA damage associated with cancer is at least a component of aging. This view is supported by the apparent correlation between maximum lifespan and DNA repair capability seen in species comparisons — as well as by the signs of accelerated aging seen in many DNA repair diseases.
Clues about the molecular mechanisms of aging & cancer in general could be gained by comparative analysis of the mechanisms of segmental progerias leading to specific cancers & specific manifestations of aging. XP, AT & Werner's Syndrome are segmental progerias due to defective NER, defective cell cycle control & defective recombination (respectively) leading to high rates of skin cancer, leukemia & sarcomas (respectively). The cancer symptoms are more prominent with XP & AT, whereas the progeria is more prominent with Werner's Syndrome. Down's Syndrome & Hutchinson-Gilford Syndrome are segmental progerias not particularly associated with high cancer risk. Defective DNA mismatch repair leads to a form of colon cancer (HNPCC) without symptoms of accelerated aging.
What is the relative contribution of reduced vulnerability to cancer due to reduced Insulin-like Growth Factor−1 (IGF−1) to the extended lifespan of dwarf mice and to what extent or by what mechanism is the rate of aging slowed?
Dietary factors, smoking and environmental chemicals can play a significant role in the incidence of cancer, as indicated by the fact that breast cancer in North American women is ten times more common than for women in Japan. And dietary antioxidants — if not supplemental — appear to reduce the risk of cancer. Environmental factors associated with aging or maximum lifespan might cause increased glycation, generalized macromolecule damage and lipofuscin accumulation along with DNA damage.
But in the absence of other diseases, there is a general and exponential increase in the likelihood of contracting cancer as a subject (human or other mammal) ages. There is an increased cumulative effect of DNA mutation and a decline in immune-system function with age. Nonetheless, the pattern of cancer increase associated with aging is very different from immune deficiency disease. Whales have 600 times as many cells as humans yet suffer no greater incidence of cancer. It is improbable that whales have an immune system that is 600 times better than that of humans. Whales must have other special defenses against cancer (which would be well worth learning to understand). The high rate of cancer in rodents is not surprising in light of the proclivity to immortalization associated with their telomeres. But the capacity of mammalian species to detoxify the carcinogenic chemical benzo(a)pyrene to a water-soluble form also correlates well with maximum lifespan [EXPERIMENTAL CELL RESEARCH 116:359-364 (1978)].
DNA damage due to mutagens more readily leads to cancer, but defective DNA repair more readily leads to aging. Nearly all of the "accelerated aging" diseases involve defective DNA repair. Better DNA repair allows the deer mouse to live much longer than the house mouse. It may be that mutagens damage both DNA as well as cellular defenses against DNA damage, but that when DNA repair is defective cells can respond by inducing cellular senescence or apoptosis — preventing cancer, but accelerating aging. With aging the declining efficiency of cellular mechanisms means that there is a decreasing likelihood that cancerous cells will be eliminated by apoptosis.
For technical details about the nature of cancer (and methods of prevention) — see my essay Cancer Death.
Individuals of different species seem to age at different rates for different reasons. Laboratory studies of lifespan is currently only feasible for short-lived species, but if some biomarker could be found for determining biological age (rather than chronological age) then human lifespan studies would be feasible. A biomarker of aging would be a better predictor of life expectancy and future functionality than chronological age. Unfortunately, we even lack a method for biomarker validation. And if a biomarker could be validated for rodents, how could we prove that the biomarker applied equally-well to humans? Without biomarkers of aging we cannot say definitively if "accelerated aging" diseases exist.
Without validated biomarkers of aging, it is difficult to prove that nutrients, drugs or other interventions are slowing aging and extending the maximum lifespans of humans. With biomarkers, it would only be necessary to show reduced deterioration within a reasonable time-frame (a few years) in humans. Without biomarkers, positive proof of an anti-aging intervention for humans could only come by observing effects on lifespan in studies lasting decades or centuries. To be of use within our own lifetimes, the results from short-lived mammals may be the best we can hope for if biomarkers are not found. Despite years of effort, biogerontologists have not had much success in their search for biomarkers of aging [EXPERIMENTAL GERONTOLOGY; Johnson,TE; 41(12):1243-1246 (2006) and BIOLOGICAL CHEMISTRY; Simm,A; 389(3):257-265 (2008)].
Insofar as Caloric Restriction with Adequate Nutrition (CRAN) seems to slow aging in rodents and many other short-lived species, long-term studies of CRAN on monkeys are being conducted to establish if CRAN also slows aging in primates. Although it will take decades for these studies to run to completion and current data is not yet statistically significant, rhesus monkeys on CRAN show the same reductions of body temperature & plasma insulin as CRAN rodents, as well as showing a slower decline in serum DeHydroEpiAndrosterone Sulfate (DHEAS). Men with greater survival in the Baltimore Longitudinal Study of Aging also show reduced body temperature & plasma insulin, along with elevated serum DHEAS — suggesting that these three factors may be biomarkers of biological age [SCIENCE 297:811 (2002)].
Skin biopsies from CRAN & control nonhuman primates have been used to assess glycation & glycoxidation (oxidation of glycation products to form AGEs). Furosine as a measure of glycation increased mildly with age in the control animals and this increase was significantly reduced in CRAN animals. Using pentosidine as a measure of glycoxidation, no significant variations were observed — but results for tissues other than skin might have been different [JOURNALS OF GERONOTOLOGY 58A(6):508-516 (2003)].
F2−isoprostanes are stable products of oxidized arachidonic acid which can be readily measured in urine to quantify lipid peroxidation. Plasma concentrations rise dramatically with age in rats, providing support for the association of lipid peroxidation with aging and for the potential of F2−isoprostanes as biomarkers of aging [BIOCHEMICAL AND BIOPHYSICAL RESEARCH COMMUNICATIONS 287:254-256 (2001)]. (For details on isoprostanes see Essential Fatty Acids in Cell Membranes.)
A statistically significant trend (with extremely wide variation) of T−cell subsets in mice provides a potential biomarker. Mice with low levels of CD4 & CD8 memory cells, high levels of CD4 naive cells and low levels of P−glycoprotein CD4 cells live 6% longer [JOURNALS OF GERONTOLOGY; Miller,RA; 56A(4):B180-B186 (2001)]. The "biomarker" based on these four T−cell subsets was able to predict longevity of 18-month-old mice with P−values less than 0.003. Mice most often die of cancer, so this might be a better indicator of mortality risk. But if valid, would this biomarker indicate that the immune system theory of aging predominates, or would it indicate that the aging process simply impinges most predictably on the immune system?
With aging the sleeping EEG patterns known as "sleep spindles" and "K−complexes" diminish in number — and it has been suggested that this change can be used as a biomarker of brain aging [CLINICAL NEUROPHYSIOLOGY; Crowley,K; 113(10):1615-1622 (2002)]. There is also reduced circadian signaling with age, and much of this reduction may be due to reduced melatonin secretion [NEUROBIOLOGY OF AGING; Munch,M; 26(9):1307-1319 (2005)].
In the Framingham study (a longitudinal epidemiological study of large size and long duration in Framingham, Massachusetts that has focused on cardiovascular disease risk factors) lung volume (largest volume of air that can be voluntarily expelled from the lung) — which decreases with age in smokers & non-smokers — was well correlated with risk of death in the 45−74 year-old age-range. But even lung volume was inferior to chronological age as a predictor of overall mortality risk. Forced expiratory volume in one second remains the best predictor of all-cause mortality [CHEST; Schunemann,HJ; 118(3):656-664 (2000) and EUROPEAN RESPIRATORY JOURNAL; Young,RP; 30(4):616-622 (2007)], but that does not mean that it is a biomarker of aging.
If mortality risk were the definitive characteristic of aging, then standing in an open field criss-crossed with machine-gun fire would be a biomarker of aging. If aging is damage to organs, tissues, cells and macromolecules then many kinds of damage need to be considered. Certain kinds of damage are more related to specific disease conditions than generalized "aging". Damage to substantia nitra cells leads to Parkinson's Disease, nuclear DNA mutations lead to cancer, glycation of lens crystallins leads to cataracts, etc. Nonetheless, aging increases the predisposition to these disease conditions.
Partly because of the failures to find biomarkers, some biogerontologists question that a unitary process of aging exists — asserting that the phenomenon called "aging" is really multiple degenerative processes operating in parallel. A unitary cause of aging might necessitate discovery of a unitary biomarker. The multiple forms of damage to macromolecules, cells and tissues associated with aging points to multiple causes, and would necessitate multiple biomarkers. Despite the fact that different mechanisms must be involved, the rather uniform slowing of aging seen for dwarf mice and CRAN-diet (versus ad-libitum fed) animals would seem to validate the existence of a unitary aging process — as does the comparison of aging rates between species or breeds of dogs.
But aging can only be the result of damage to macromolecules: proteins, lipids, carbohydrates, and DNA (including telomeres). Causes of aging damage are reactive oxygen & nitrogen species, sugars (glycation), radiation, pathogens, inflammatory cytokines, and accumulated toxins (metals, PCBs, dioxins, etc.). The different aging rates of different species is due to the fact that endogenous damage is produced at different rates (eg, bird mitochondria produce fewer free radicals than mammalian mitochondria), different protective mechanisms exist (eg, naked mole rats arrest cancer growth with contact inhibition), and more long-lived species more effectively eliminate damage (eg with better lysosome enzymes, better DNA repair, better autophagy, etc.). If aging is programmed genetically, it can only be programmed to reduce damage formation or remove/repair damage better or worse.
Exogenous agents can accelerate forms of aging damage. Diabetes and dietary
Advanced Glycation End-products (AGEs) accelerate protein
cross-linking [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Koschinsky,T;
94(12):6474-6479 (1997)]. Immunosenescence is substantially associated with
cytomegalovirus [CURRENT OPINION IN LIPIDOLOGY; Derhovanessian,E; 21(4):440-445
(2009)]. White blood cell telomere attrition is accelerated in obesity and insulin
resistance [CIRCULATION; Gardner,JP; 111(17):2171-2177 (2005)]. There is
considerable overlap in the histopathology of skin photoaging and skin intrinsic
aging [EXPERIMENTAL DERMATOLOGY; El-Domyati,M; 11(5):398-405 (2002)].
A high fat meal elevates plasma inflammatory cytokines more than a high carbohydrate
meal [JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY; Nappo,F; 39(7):1145-1150
(2002)], and plasma inflammatory cytokines are substantially associated with
age-related cataract [AMERICAN JOURNAL OF OPTHALMOLOGY; Klein,BEK; 141(1):116-122 (2006)].
F2−isoprostanes (the best marker of lipid peroxidation) are
substantially elevated in the foam cells of atherosclerotic
plaque [JOURNAL OF CLINICAL INVESTIGATION; Pratico,D; 100(8)2028-2034 (1997)],
and F2−isoprostanes in the urine of smokers drops by
more than one third after two weeks of smoking
cessation [NEW ENGLAND JOURNAL OF MEDICINE; Morrow,JD; 332(18):1198-1203 (1995)].
Because aging is due to multiple forms of damage, there can be no
singular underlying biological age. Cause of death and impairment of
functionality will be a function of which form of damage is the
greatest — which will vary from person to person. Rather than
engage in a fruitless search for a biological age (biomarker of aging),
biogerontologists should seek assays for every possible form of aging
damage. Damage assays can allow for ranking forms of aging damage,
prioritizing interventions, and monitoring intervention effectiveness.
Caloric Restriction with
Adequate Nutrition (CRAN) dramatically extends the maximum lifespan of laboratory animals.
Victims of starvation & malnutrition are not experiencing the life-extending
benefits of CRAN — adequate nutrition (vitamins, minerals, essential
amino acids and essential fatty acids in adequate quantity) is absolutely necessary
for calorie restricted diets to extend lifespan. Because almost every aspect of the
aging process appears to be slowed by CRAN, studying CRAN has become a means of
defining & understanding the aging process itself — including the search for
biomarkers of aging.
Rats, mice and hamsters experience maximum
lifespan extension from a diet which contains 40−60% of the calories (but all
of the required nutrients) which the animals consume when they can eat
as much as they want. Mean lifespan is increased up to 65% and maximum lifespan
is increased up to 50%, when CRAN is begun just before puberty. Except for the puberty
effect, it is as if all animals are allotted a lifetime supply of food — and those
who eat more slowly live longer because it takes longer to consume all the food.
The mechanism by which caloric restriction has such dramatic effects is
unproven, but maturity, thymus shrinkage, DNA-repair decline and tumor
formation is delayed. The experimental animals show more complete oxidation
of fatty acids, with fewer ketones (R'RC=O) in the blood, and cell membranes have less
cholesterol & saturated fatty acids. Collagen cross-linking occurs more
slowly in rats on CRAN which have blood glucose levels reduced about 15%
below controls. Reduction of visceral body fat is associated with reduced
insulin resistance due to
reduced levels of proinflammatory cytokines [EUROPEAN JOURNAL OF CLINICAL
INVESTIGATION 32(Suppl 3):24-34 (2002)].
Oxidative damage (8−oxodG) to mtDNA is 16 times greater than to nDNA in the livers of
6−month old rats [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Richter,C;
85(17):6465-6467 (1988)]. Although CRAN does not reduce oxidative damage (8−oxodG) to
rat liver nDNA and only reduces oxidative damage to mouse liver nDNA by 19%, it completely
eliminates mtDNA damage in both the rat &
mouse [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Hamilton,ML;
98(18):10469-10474 (2001)]. One year of CRAN in rats has been shown to reduce liver
mitochondrial hydrogen peroxide production from Complex I by 47% [FREE RADICAL
BIOLOGY & MEDICINE; Lopez-Torrez,M; 32(9):882-889 (2002)]. For mammalian species, a
negative exponential correlation has been demonstrated between liver mitochondrial hydrogen
peroxide production and maximum lifespan [JOURNAL OF COMPARATIVE PHYSIOLOGY B;
Perez-Campo,R; 168:149-158 (1998)].
Although CRAN animals produce fewer free radicals, their metabolic rate
(oxygen consumption per gram of tissue) is not reduced. The inner mitochondrial membranes
of CRAN animals have a higher saturated/unsaturated fat ratio making them less vulnerable
to proton leak from lipid peroxidation. Both state 3 & state 4 respiration rates
are greatly reduced in brain, heart & kidney tissue [THE INTERNATIONAL JOURNAL
OF BIOCHEMISTRY & CELL BIOLOGY 34:1340-1354 (2002)]. CRAN rats show 15% less plasma
glucose and 50% less plasma insulin than controls, having the same rate of glucose
utilization per unit mass, meaning that glucose is being more efficiently
utilized [JOURNAL OF GERONTOLOGY; Masoro,EJ; 47(6):B202-B208 (1992)].
Macroautophagy is normally induced during conditions of starvation. CRAN
in the nematode C. elegans induces macroautophagy, whereas
inhibiting the genes required for macroautophagy inhibits the genes require
for autophagy — and prevents CRAN from extending
lifespan [PLOS GENETICS; Hansen,M; 4(2):e24 (2008)].
A convincing case has been made that CRAN operates by
evolutionarily-conserved mechanisms of nutrient-sensing molecular
pathways (insulin/IGF-1) in yeast, worms, flies, and
mammals [SCIENCE; Fontana,L; 328:321-326 (2010)]. CRAN does not
increase insulin sensitivity or extend the lifespan of Growth-Hormone Receptor
Knock-out mice, suggesting that insulin sensitivity plays a key role
in life extension by
CRAN [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA);
Bonkowski,MS; 103(20):7901-7905 (2006)]. Exercise, however, increases
insulin sensitivity without increasing maximum
lifespan [AGE; Fontana,L; 32(1):97-108 (2010)].
Reduced body size within a species often correlates with longer lifespan
and reduced plasma IGF−1. Great Danes (400 ng/mL plasma
IGF−1) live about 7 years, whereas
Chihuahuas (40 ng/mL plasma IGF−1) can live over 15 years.
Insulin increases IGF-1 activity by lowering serum IGF-binding
protein [JOURNAL OF BIOLOGICAL CHEMISTRY; Powell,DR;
266(28):18868-18876 (1991)].
Longevity in humans is correlated with a genetic predisposition
to low plasma IGF-1 [JOURNAL OF CLINICAL ENDOCRINOLOGY & METABOLISM;
Bonafe,M; 88(7):3299-3304 (2003)]. Rodents consistantly show reduced
plasma IGF-1 on CRAN, but for humans plasma IGF-1 is reduced by protein
restriction, not calorie
restriction [AGING CELL; Fontana,L; 7(5):681-687 (2008)].
An ongoing study of rhesus monkeys (which have a maximum lifespan of 40 years)
has shown a 50% reduction in cancer and cardiovascular disease for the animals
subjected to 30% calorie restriction compared to
controls [SCIENCE; Colman,RJ; 325:201-204 (2009)].
But the rhesus monkey studies are still in
progress [SCIENCE; Roth,GS; 297:811 (2002)].
There is reasonable evidence that the benefits of CRAN seen in rodents apply to humans.
Contrary to former reports of a "J−shaped" relationship between body
weight and human mortality, when corrections are made for smoking and underlying
disease the relationship is linear [NEW ENGLAND JOURNAL OF MEDICINE; Manson,JE;
333(11):677-685 (1995)]. Of course, anorexics who are malnourished for micronutrients
are not examples of human CRAN. There may well simply be a continuum between
CRAN and the metabolic syndrome, meaning the benefits of CRAN are
simply a matter of quantity of calories, versus the idea that CRAN is
some qualitatively distinct metabolic state.
Humans who have practiced CRAN for about six years show considerable
reduction in risk factors for atherosclerosis, including reduced
LDL-cholesterol, increased HDL-cholesterol, reduced serum triglycerides,
and reduces systolic and diastolic blood
pressure [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA);
Fontana,L; 101(17):6659-6663 (2004)]. It is also to be expected that
CRAN reductions of inflammatory cytokines and growth factors would result
in reduced cancer in humans, as has been seen in rodents and
monkeys [TRENDS IN PHARMACOLOGICAL SCIENCES; Longo,VD; 31(2):89-98 (2010)].
Elderly human subjects
(60 years average age) restricting calories 30% for 3 months showed
a 20% increase in verbal memory
scores [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Witte,AV;
106(4):1255-1260 (2009)].
Substantial evidence indicates that as much as half of the
life-extension benefits of CRAN are due to restriction of the single amino acid
methionine.
The lifespan of Drosophila fruit flies can be extended by reducing
casein or methionine [NATURE; Flatt,T; 462:989-990 (2009)]. In a study of rats given
20% the dietary methionine of control rats, mean
lifespan increased 42% and maximum lifespan increased
44% [THE FASEB JOURNAL;Richie,JP; 8(15):1302-1307
(1994)]. A study of male rats subjected to methionine restriction,
but no restriction of calories, showed the same decreases in mitochondrial reactive oxygen
species and oxidative damage to DNA as was seen with rats subjected to
CRAN [FASEB JOURNAL; Sanz,A; 20(8):1064-1073 (2006)].
(See Life Extension
Benefits of Methionine Restriction for more details.)
Attempts have been made to find a pill that would mimic the effects of CRAN. Using
gene chips it has been found that Metformin results in the same gene expression seen in CRAN. (See
http://www.lef.org/magazine/mag2003/2003_preprint_bio_01.html
and
http://www.lef.org/magazine/mag2001/sep2001_report_metformin_01.html.) But Metformin only
extends mouse lifespan a third as much as CRAN.
For technical details about CRAN — plus an account of my personal
experiences with CRAN — see my essays
Caloric Restriction with Adequate Nutrition — Overview ,
My Practice of Caloric Restriction with Adequate Nutrition
and My Current Health Regimen — Exercise, Diet, Supplements.
As I stated in the beginning, my primary concern in this essay has been
to elucidate the mechanisms of aging, rather than
methods to prevent it. But the goal
of my understanding is indeed to apply
an understanding of mechanisms to the evaluation of methods. Literature
on methods to extend maximum lifespan which is strongly
grounded in scientific research is rare. The primary reason for this
scarcity is lack of funding for such research.
In contrast with maximum lifespan, there is a vast
amount of literature on ways to extend mean (average)
lifespan through diet & exercise and avoidance of dangers, toxins
& disease. Only 26% of smokers live to age 80, in contrast with
57% of nonsmokers [ADDICTION 97:15-28 (2002)]. A practical
life-extensionist currently has far more to gain by utilizing
information available on extending mean lifespan than by preoccupation
with maximum lifespan. Some misguided life-extensionists have discounted
the use of anti-oxidant supplements because they have only been shown
to be of benefit in extending mean lifespan, not maximum lifespan.
A prime candidate for a biomarker
of aging (which has been a focus of attention in the calorie restriction with
adequate nutrition studies of primates) has been
insulin resistance.
Reduced glycation may be achieved by reduction of typical blood glucose
levels. Low fat meals are one means to achieve this because fatty acids
promote insulin resistance — and greater insulin resistance means that
higher blood glucose levels are required to supply cells with the same
amount of glucose (causing more glycation) [THE NEW ENGLAND JOURNAL OF
MEDICINE 342(19):1440-1441 (2000) and DIABETES CARE 20(11):1774-1780 (1997)].
(Insulin resistance is a fundamental cause of adult-onset diabetes.) Also,
increased consumption of soluble fiber (particularly the beta-glucan found
in oat bran & barley) lowers 24−hour plasma glucose & insulin
concentrations. Experiments demonstrating that lysine-glycation predicts early
death in both CRAN & freely-fed rats makes lysine-glycation a very promising
biomarker candidate [FASEB JOURNAL 14:145-156 (2000)]. (For more on these
subjects see
the metabolic
syndrome)
Although no substance has
been shown conclusively to extend maximum lifespan in humans, a few studies
indicate that some supplements may extend the lives of laboratory mammals
(mice, rats or guinea pigs, usually). The are quite a few studies indicating
that Deprenyl, for example,
has extended the maximum lifespan of a variety of mammals.
There is at least one book (self-published), which is based on a serious
attempt to search the scientific literature for methods to extend
maximum lifespan.
Dr. Thomas Donaldson has reviewed those supplements that appear to extend
the lifespan of mammals in at least one scientific study
in his self-published book A GUIDE TO ANTI-AGING DRUGS. It
would be more accurately titled A GUIDE TO ANTI-AGING SUPPLEMENTS because,
although Procaine, Deanol,
Deprenyl,
Levodopa, Phenformin and Phenytoin
deserve to be called drugs, Vitamin E, Pyridoxine, Pantothenate, Melatonin,
Cysteine, Chromium and Coenzyme Q10 do not. Five mechanisms are identified
by which these supplements work:
(1) anti-oxidation
Dr. Donaldson died early in 2006 and his self-published book
may be difficult to obtain.
Although Hormone Replacement Therapy (HRT) to bring androgens, estrogens and
growth hormone to youthful levels improve cognitive function & muscle tone (among other
benefits) these hormones promote cancer growth and therefore may be dangerous to use until
cancer is preventable & curable. By contrast,
DHEA not only protects against obesity,
diabetes & autoimmune disease, it reduces cancerous tumor-formation [ADVANCES
IN ENZYME REGULATION 26:355-382 (1987)] and can protect against excitotoxic damage in
the hippocampus [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA) 95(4):1852-1857
(1998)].
The nutraceuticals section of my website describes a number of supplements
which may extend average, if not maximum, lifespan.
Life
Extension Magazine — which is available at no cost online — regularly
publishes articles with many fine citations to research on nutraceuticals
(primarily animal studies) which could potentially be of great benefit in
extending human life.
Cardiovascular function declines and insulin resistance increases
with age for most people. But most of these changes can actually be
attributed to declining physical activity and increasing abdominal
obesity associated with aging — rather than senescence per
se. Higher HDL-cholesterol, lower triglycerides, lower
insulin-resistance & prevalence of diabetes, good cerebral
perfusion, good glucose metabolism, good
cardiovascular function and good endocrine function of older people
who engage in regular vigorous aerobic exercise are nearly to the
level of that seen in their younger counterparts — in sharp
contrast to their sedentary peers [NEW ENGLAND JOURNAL OF MEDICINE
328:533-537 (993); AMERICAN JOURNAL OF PHYSIOLOGY 268:E484-E490
(1995); JOURNAL OF THE AMERICAN GERIATRIC SOCIETY 38:123-128 (1990)].
Health problems caused by sedentary living are too often blamed on
senescence.
Exercise is well-known to lower blood pressure and otherwise improve
cardiovascular health. And, as has been mentioned, exercise can boost
rejuvenating Growth Hormone (GH) more effectively than injections.
Exercise also normalizes other hormone responses to youthful levels
[JOURNALS OF GERONTOLOGY:BIOLOGICAL SCIENCE 51A(1):B30-B37 (1996)] and
reduces insulin resistance [METABOLISM 44(10):1259-1263 (1995)] while improving immune
function [MECHANISMS OF AGING AND DEVELOPMENT 93:215-222 (1997)].
Although excessively strenuous exercise can generate harmful levels of free
radicals, regular endurance exercise protects against free radicals by increasing
muscle levels of SuperOxide Dismutase (SOD), glutathione peroxidase
and reduced glutathione (GSH) (but has no effect on catalase)
[MEDICINE & SCIENCE IN SPORTS & EXERCISE 31(7):987-997 (1999)].
Vitamin E is particularly protective against exercise-induced
free radicals [AMERICAN JOURNAL OF PHYSIOLOGY 264:R992-R998 (1993)].
Vitamin E has a pro-oxidant potential that can only be prevented
by agents like Vitamin C and CoEnzyme Q10, which eliminate the
alpha-Toc. radical [ARTERIOSCLEROSIS, THROMBOSIS
AND VASCULAR BIOLOGY 16:687-696 (1996)]. (For more on exercise, see
Exercise)
According to the Honolulu Heart Program, the best predictors of "successful
aging" were low blood pressure, low blood sugar, abstinence from tobacco and
not being obese. The Framingham study concluded that
by holding 11 different risk factors (such as blood pressure & serum
cholesterol) at the 30-year-old level, women would live to be 97 and
men would live to age 100. As the above review should indicate, many of
the afflictions of aging (including vascular dementia) are the result of
poor cardiovascular health. Therefore, despite the fact that maximum
lifespan is not extended, the effects of extended youth & extended health
would nonetheless be expected from measures extending average lifespan —
cardiovascular health, in particular. Atherosclerosis not only increases
blood pressure and the risk of death from stroke & heart attack, but
reduces the health & function of all organs (including the brain)
through impaired circulation.
It is difficult to
gain much immediate benefit from insights into molecular mechanisms of
aging, but enormous immediate benefit can be gained from reducing
calorie intake (while maintaining adequate nutrition), avoiding tobacco,
avoiding alcohol,
exercising, taking supplements, eating low-fat/high-fiber diets, etc.
Epidemiological evidence indicates that adherence to a vegetarian diet for more
than two decades can increase lifespan 3.6 years [AMERICAN JOURNAL
OF CLINICAL NUTRITION 78(Suppl):526S-532S (2003)].
And cryonics may serve
as "first-aid" to transport us to the time when significant
advances in the elimination of senescence have occurred.
Some geronotologists believe that somatic gene therapy
can accomplish such goals as removing the telomerase gene from somatic cells (to reduce
cancer), migrating mitochondrial DNA into the nucleus and utilizing bird mitochondria genes
to create modified human mitochondria which produce fewer free radicals. With
"adequate funding" these gerontologists believe an ageless
mouse can be created within a decade
(The Methuselah Mouse Prize).
Every year we can add to our lives now increases our chances of
living to the time when technology can eliminate & reverse aging — or
cryonics can induce perfect suspended animation.
This essay is not the place to summarize every practice that can
possibly extend life or delay/avert death. See the pages on this
website dealing with
Health,
Nutraceuticals,
Life-Extension,
CRAN,
Cryonics,
Death by Murder, and my
statistical summary of all causes of death .
A healthy lifestyle, CRAN,
and perhaps even supplements can do no more than slow the aging process or
extend mean lifespan. Enduring youth might be attained if aging could be stopped
at a youthful age, but it seems unlikely that the damage to organs, tissues, cells
and molecules known as aging can be stopped completely. Replacing or repairing
damaged organs, tissues, cells and even molecules seems like a better strategy. These
strategies can restore function to old organisms — can even rejuvenate.
Replacement of old or defective organs is a regenerative technique which has been
tantalizingly close for decades. Only a small fraction of potential candidates for
heart, kidney or liver transplants are able to benefit, because of low availability
and immune incompatibility. The development of a completely mechanical heart remains
out of reach, but there is hope that ventricular assist devices supporting the
left ventricle could benefit most end-stage heart-disease patients. Pigs have many organs
whose size is compatible for human transplant, but immune compatability and the threat
of viral infection remain obstacles.
Although the liver can mostly regenerate lost tissue, wounds to most
body tissues (including myocardial infarction) result in scar formation
rather than regeneration of functional tissue. Stem cells could allow
for true tissue regeneration. Human
Embryonic Stem Cells (ESC) have the
greatest potential to differentiate into any desired tissue type.
Retrovirus
induction of overexpression of certain proteins can generate
induced Pluripotent Stem Cells (iPSC)
from fibroblasts. iPSC are nearly as pluripotent as ESCs. But both
ESCs and iPSCs can form
teratomas
(benign tumors) and induce antigenic tissue rejection (although iPSCs
are less antigenic than ESCs). Antigenicity can be reduced or eliminated
by regenerating the
thymus
gland [NATURE; Chidgey,AP; 453:330 (2008)], by such
means as androgen
blockage> [THE JOURNAL OF IMMUNOLOGY; Sutherland,JS; 175(4):2741-2753 (2005)].
When available, adult stem cells from the target tissue of the afflicted
patient are ideal for avoiding an antigenic response. But too often (as in
cases of tissue degeneration) such stem cells are not available. Stem cells
from the
umbilical cord
cryogenically stored at birth have the potential for tissue regeneration
later in life.
Most attempts at genetic repair have traditionally involved the use of a retrovirus
to insert a new gene into a random position on a chromosome. But by attaching
zinc fingers (which determine
where transcription factors bind) to endonucleases (which break DNA strands) homologous
recombination can be induced to correct and replace defective (or undesired) DNA
sequence. The first applications of this technology are to isolate stem cells from
the bone marrow of patients having blood disease mutations, to correct those
mutations in lab dishes using zinc finger nucleases and to transplant the stem cells
back into the patients [SCIENCE; 310:1894-1896 (2005)].
Regenerative medicine looks for means to mimic salamanders (which can regrow
severed limbs), newts (which can regrow not only limbs, but intestine, jaw and spine)
and zebrafish (which can regrow a heart) — by replacing the dead scar tissue after
a heart attack with new heart cells.
Regenerative medicine uses three different strategies: (1) implantation
of stem cells from culture into an existing tissue structure (2) implantation of
stem cells into a tissue scaffold that guides restoration or (3) induction of
residual cells of a tissue structure to regenerate the necessary body part. A
salamander can not only regenerate a limb, but can regenerate the lens or retina
of an eye and can regenerate an intestine. For regeneration the salamander tissues
form a blastema by dedifferentiation of mesenchymal cells, and the blastema
functions as a self-organizing system to regenerate the
limb [SCIENCE; 310:1919-1923 (2005)]. DNA microarray analysis of salamanders has shown that humoral immune
and local tissue factors control the initial phase of limb regeneration, but nerve-derived
factors later become crucial [BMC BIOLOGY; Monaghan,JR; 7:1-19 (2009)].
The MRL mouse, unlike other mice, can regenerate
damaged heart muscle without scar
formation [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Leferovich,JM;
98(17):9830-9835 (2001)]. Regenerative medicine would also aim to replace substantia
nigra cells in Parkinson's Disease and regrow a spinal cord after spinal cord injury.
Multipotent adult progenitor cells, such as bone marrow cells, have been shown to be
capable of replacing myocardial tissue destroyed by ischemic heart
disease [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Orlic,D;
98(18):10344-10349 (2001)].
Senescent cytotoxic T-cells have been removed from the serum of mice by
attachment of iron oxide nanoparticles linked to antibodies and applying
a magnetic field to the serum in an extracorporeal circuit [REJUVENATION
RESEARCH; Rebo,J; 13(2-3):298-300 (2010)].
Injured skeletal muscle has the capacity to regenerate in young mammals, but this
capacity is considerably impaired with aging. Activation & proliferation of muscle
regenerating progenitor cells (satellite cells) is dependent upon signalling from
transmembrane Notch receptors. Notch receptors have several ligands, ie,
extracellular molecules that the receptor requires to function. Upregulation of the
Notch ligand Delta has been shown to be sufficient to restore the regenerative
potential of skeletal muscle in old mice [SCIENCE; Conboy,IM; 302:1575-1577 (2003)],
Caution is advised in upregulating Notch, because overexpression of Notch can lead to
cancer [BREAST CANCER RESEARCH; Dontu,G; 6:R605-R615 (2004)]. The blood
plasma of young mice have been reported to restore the regenerative potential of both
muscle and liver cells in old mice [NATURE; Conboy,IM; 433:760-764 (2005)]. High
levels of TGF−ß (Transforming Growth Factor beta) in the blood of old mice
appears to be the problem. Systemic (serum) TGF−ß is
immunosuppressive [THE JOURNAL OF EXPERIMENTAL MEDICINE; Wahl,SM; 180(5):1587-1590 (1994)]
and aged cells have been shown to produce increased levels of
TGF−ß [IMMUNOLOGY LETTERS; Zhou,D; 36(1):7-12 (1993)]. Muscle regeneration
normally makes use of inflammatory
processes [AMERICAN JOURNAL OF PHYSIOLOGY; Tidball,JG; 288(2):R345-R353 (2005)]
and TGF−ß has been shown to inhibit muscle
regeneration [CIRCULATION RESEARCH; Zhu,S; 94(5):617-625 (2004)].
Organ transplant or even tissue transplant would not be of much benefit for an
aging brain, which is composed of non-dividing, enduring cells (neurons) whose
continued existence is crucial for the retention of knowledge and identity. In this case,
rejuvenation could be done on a molecular level rather than at the tissue or organ level.
For example, Aubrey de Grey has suggested that genes taken from bacteria could be
transmitted into the genome of human neurons to produce enzymes that dissolve &
eliminate lipofuscin, thereby rejuvenating the neuron. The same gene in blood vessel
"foam cells" could reverse atherosclerosis. There is evidence that the
extracellular protein cross-linking due to glycation which
leads to arterial wall stiffening as well as stiffening of the left ventricle can be
reversed by the thiazolium derivative ALT−711, which catalytically breaks
cross-links [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA)
97(6):2809-2813 (2000) and
CIRCULATION; 104(13):1464-1470 (2001)].
Biogerontologist Aubrey de Grey believes that reversing
aging may actually be more feasible than slowing aging, in the same sense that is sometimes
more economical to periodically repair damage than to go to extraordinary expense to
slow the rate of damage. Dr. de Grey believes that the key to rejuvenation is the
repair of seven distinct kinds of damage that represent aging: cell loss, cell senescence,
extracellular protein cross-linking, nuclear DNA mutations, mitochondrial DNA mutations
and the accumulation of garbage inside cells as well as outside cells. He has characterized
the repair of these seven kinds of damage as
Strategies for Engineered Negligible Senescence (SENS).
The seven repair strategies that Dr. de Grey advocates can
be summarized: (1) Cell loss can be repaired (reversed) just by suitable exercise
in the case of muscle, but for other tissues it needs various
growth factors to stimulate cell division, or in some cases it
needs stem cells. (2) Senescent cells
can be removed by activating the immune system against them. Or
they can be destroyed by gene therapy to introduce "suicide genes"
that only kill senescent cells. (3) Protein cross-linking can
largely be reversed by drugs that break the links. But for some of the
links we may need to develop enzymatic methods. (4) Extracellular
garbage can be eliminated by vaccination that gets immune cells
to "eat" the garbage. (5) For intracellular junk we need to
introduce new enzymes, possibly enzymes from soil bacteria, that
can degrade the junk that our own natural enzymes cannot degrade.
(6) For mitochondrial mutations the plan is not to repair them but
to prevent harm from the mutations by putting suitably modified copies of
the mitochondrial genes into the nucleus by gene therapy. The
mitochondrial DNA experiences so much mutation damage because
most free radicals are generated in the mitochondria. If
mitochondrial DNA can be moved into the nucleus it will be
better protected from free radicals, and there will be better
DNA repair when damage occurs. All mitochondrial proteins would
then be imported into the mitochondria. (7) For cancer (the
most lethal consequence of mutations) the strategy is to use gene
therapy to delete the genes for telomerase and to eliminate
telomerase-independent mechanisms of turning normal cells into
"immortal" cancer cells. To compensate for the loss of telomerase
in stem cells we would introduce new stem cells every decade or so.
For more background on Dr. de Grey's approach, see
SENS Overview.
The ultimate rejuvenation, however, will occur further in the future with the advent
of molecular repair technology (nanotechnology) which can fix all kinds of
molecular damage due to aging (as detailed in the book
ENGINES OF CREATION by
K. Eric Drexler).
What causes aging? In other words, what lies behind the progressive deterioration that
accompanies the passage of time after maturity — with special interest to humans. To
answer in outline form:
An organism that can create fewer free-radicals in generating energy (more efficient
mitochondria), use less energy to live, have more effective antioxidant defenses, have
better DNA protection, have better DNA repair, have a better immune system and detoxify
more effectively in the liver — can reduce damage from endogenous & exogenous sources.
Glucose is necessary for energy production, but glucose causes glycation of proteins.
Energy creation results in free radicals as a toxic byproduct. Toxic & non-toxic
garbage accumulation is primarily a problem for non-dividing cells (like neurons &
muscle cells) which cannot dilute-away the garbage. The damage which causes
aging is the damage due to necessary metabolism. This damage affects DNA repair, antioxidant
production, telomere length, cell-cycle control, proteosome function, etc. — resulting in
reduced capacity to cope with increasing levels of damage.
Telomere shortening contributes to mortality only in a few tissues. Neurons & muscle
cells are non-dividing and are thus not affected by telomere shortening. Telomere
shortening may contribute to mortality most significantly for immune system cells
& arterial epithelial cells. Even if telomere shortening in the immune system is proven
to cause the majority of deaths in the very elderly, the mortality is better described as
"failure of the weakest link" (like the death of wild horses from worn-down
teeth) than as aging. If biological gerontologists are successful in finding
means to greatly increase human lifespan, then telomere shortening in proliferative
tissues may become far more relevant to human aging. (For non-dividing cells, notably
neurons, metabolic damage & garbage accumulation could be considered the "weakest
link" if it weren't for the fact that cell death is so different from cell
senescence.)
Metabolic damage would be much less of a problem if its byproducts (cross-links, AGEs,
lipofuscin, etc.) could be eliminated — along with whatever toxins (lead, cadmium, DDT,
PCBs, etc.) manage to enter the organism. The so-called immortality of germ cells, bacteria
and Hydra [EXPERIMENTAL GERONTOLOGY 33(3):217-225 (1998)] is probably due to
the diluting-away of toxins (all of the Hydra cells are dividing cells).
Lobsters — which have been proposed as candidates for negligible senescence
— discard tissue by molting and appear to continue growing without ever maturing. Lobsters
express telomerase in all organ tissues and may avoid senescence by the same mechanism as
Hydra [FEBS LETTERS; Klapper,W; 439(1-2):143-146 (1998)].
Why does CRAN (Caloric Restriction with
Adequate Nutrition) extend lifespan? The most plausible explanation is that the lower
level of calorie utilization & energy production allows for lower levels of blood glucose
(less glycation) and less free radical production. Efforts to duplicate CRAN with a pill
or genetic manipulation probably have no chance of success.
If the "accelerated aging" diseases
are a guide, damage to DNA — mitochondrial (mtDNA) and nuclear (nDNA) — are the
damage that is most central to aging. Damage to nDNA and nDNA repair capability
would be the worst because mitochondria (and mtDNA) can be replaced by lysosome
recycling. But the source of that nDNA damage would still be mitochondria.
Defective nDNA repair along with associated cell senescence & apoptosis leads
more to aging, whereas the nDNA damage itself leads
more to cancer. For mtDNA damage, the damage becomes most serious when the lysosomes are
no longer capable of removing defective mitochondria which are producing high levels of
free radicals. Free radicals are the primary cause of the nDNA and mtDNA damage in the
first place. Defective mitochondria play a central role in accelerated apoptosis, leading
to tissue degradation. If defective mitochondria which produce high levels of free radicals
are the major source of aging damage, then the most effective step towards slowing aging
would be improving lysosomal function by providing more efficient enzymes to the lysosomes.
The maximum lifespan of one or a few individuals of a species is taken as a proxy for
the rate of aging of that species and for the idea that only extensions of maximum
lifespan are relevant to slow aging. But most people die of aging-related diseases:
cardiovascular disease, cancer, Alzheimer's Disease, etc. The damaging aging processes
that increase vulnerability to these diseases are more relevant to vast majority of
people than influences on maximum lifespan. For this reason it is not misleading to
speak of diabetes, tobacco, dietary AGEs, ultraviolet radiation and other exogenous
sources of tissue damage as accelerating aging — especially when the damage so
closely resembles the tissue damage normally associated with aging. For the vast
majority of people good genes can only reduce (not prevent) the aging effects of
damaging exogenous agents. (For more on this subject, see Is Longevity Entirely Hereditary?)
The "mechanisms of aging" tend to be quite tissue-specific. Replicative
senescence leads to aging of T cells and blood vessel endothelial cells, whereas
other forms of cell senescence leads to aging of stem cells in the pancreas and
selected areas of the brain. Non-mitotic cells such as neurons and myocytes are
more vulnerable to oxidative stress and DNA damage. Glycation leads to cross-linking
of extracellular proteins. For any particular individual, the combination of
heredity and environmental conditions will cause some tissues and organ systems
to age (or experience damage) more than others — and becoming the "weakest
link" leading to mortality. The number or individuals who do not succumb to
age-related death specific to a particular tissue or organ is a tiny minority.
Until molecular repair technologies are available, good health practices,
supplements and organ transplantation are our best hope of bridging
the time between now and the Age of Negligible Senescence.
To see what the elimination of aging would mean to me personally, read
my essay Why Life Extension?
XXIV. CALORIC RESTRICTION WITH ADEQUATE NUTRITION (CRAN)
XXV. OTHER METHODS TO SLOW AGING
(2) anti-glycation
(3) affecting metabolism
(4) improving the immune system
(5) acting on the brain.
XXVI. REGENERATIVE MEDICINE, STEM CELLS AND REJUVENATION
XXVII. AGING: CAUSE & CURE — SUMMARY & CONCLUSIONS
XXVIII. BOOK REFERENCES (by publication date)
(references to scientific papers are incorporated in the text)
![[GO TO BEN BEST'S HOME PAGE]](../homeback.gif) HOME PAGE
HOME PAGE