![[ Action of Secretases ]](./Abeta42.gif)
|
Alzheimer's Disease (AD) is the leading cause of dementia in the elderly and is the fourth leading cause of death in developed nations (after heart disease, cancer, and stroke), although AD victims tend to actually die of infection secondary to AD. AD affects roughly 2% of those 65 years of age, with the incidence roughly doubling every 5 years up to age 90 at which the incidence is over 50%. AD is much more prevalent in women than in men for any given age group. Estimates of percentage of dementia cases due to AD range from 50% to 80%. A Florida autopsy study of dementia victims found AD pathology in 77% of cases, whereas 26% had Lewy Body Disease, 18% had vascular dementia, 13% had hippocampal sclerosis, and 5% had FrontoTemporal Dementia [ALZHEIMER DISEASE AND ASSOCIATED DISORDERS; Barker,WW; 16(4):203-212 (2002)]. The diagnosis of AD can only be confirmed on autopsy — by the presence of amyloid plaque, neurofibrillary tangles, neuronal & synaptic loss and brain atrophy in specific brain areas. Probable diagnosis is made in a living patient (with at least 85% accuracy) on the basis of cognitive tests (especially delayed recall) and exclusion of other conditions such as stroke, hypothyroidism or nutritional deficiency.
AD is incurable. It leads to death within an average of 8 years after diagnosis, the last 3 of which are typically spent in an institution. AD is the number one cause of institutionalization in the United States. Besides memory loss, Alzheimer's patients show dramatic personality changes, disorientation, declining physical coordination, and an inability to care for themselves. In the final stages, victims are bedridden, lose urinary and bowel control, and suffer epileptic attacks. Death is usually due to pneumonia, bedsores or urinary tract infection.
For those who aspire to live a very long life, dementia is a threat second only to death — or is death in another form. If there is a possibility to prevent this disease, knowledge of how to do so should be sought vigorously. Otherwise, means to at least slow AD may provide some hope by prolonging cognitive life or buying time for future advances.
(For a more general view of the molecular biology of aging, see my monograph Mechanisms of Aging)
The molecular mechanisms and hypotheses of Alzheimer's Disease (AD) can be incredibly complex. A top-down cartoon-like overview followed by increasing depth & detail may be the best way to gain understanding.
The key event leading to AD appears to be the formation of a peptide (protein) known as amyloid beta (beta amyloid, Aß) which clusters into amyloid plaques (senile plaques) on the blood vessels and on the outside surface of neurons of the brain (amyloidosis) — which ultimately leads to the killing of neurons.
Aß peptides can cause cerebral vasoconstriction [AMERICAN JOURNAL OF PHYSIOLOGY; Niwa,K; 281(6):H2417-H2424 (2001)]. Aß has been shown to impair mitochondrial function in PC12 cells [NEUROREPORT; Pereira,C; 9(8):1749-1755 (1998)] and in human neuroblastoma cells [CELLULAR AND MOLECULAR NEUROBIOLOGY; Rhein,V; 29(6-7):1063-1067 (2009)].
A first sketch of the amyloid cascade of events in AD would therefore be:
Aß formation ⇒ amyloid plaques ⇒ neuron death ⇒ dementia
The first fact to know in creating a second sketch is that the amyloid beta peptide is created by enzyme clipping of the normal neuron membrane protein known as Amyloid Precursor Protein (APP). APP is actually thought to be a natural neuroprotective agent induced by neuronal stress or injury, which reduces Ca2+ concentration and protects neurons from glutamate excitotoxicity [NEURON 10:243-254 (1993)]. Injections of a 17-peptide subunit of APP has been shown to significantly reduce ischemic damage [EXPERIMENTAL NEUROLOGY 129:112-119 (1994)].
Enzymes can clip APP in ways that do not result in amyloid beta formation. Moreover, there
are two forms of amyloid beta peptide, one of which has 40 amino acids
and one of which has 42 amino acids. The enzymes that cleave APP are known as secretases.
The two enzymes that initially compete to cleave APP are alpha-secretase (α-secretase)
and beta-secretase (β-secretase, BACE1).
If alpha-secretase cleaves APP there is no formation
of Aß. If APP is cleaved by beta-secretase it can then be
further cleaved by gamma-secretase (γ-secretase) to form either a 40 amino acid
amyloid peptide (Aß40) which is soluble & mostly innocuous — or
a 42 amino acid peptide (Aß42) which clumps together to
form insoluble amyloid plaques. Alpha-secretase cleavage occurs at the cell surface,
whereas beta-secretase acts at the endoplasmic reticulum. Gamma-secretase produces
Aß42 if cleavage occurs in the endoplasmic reticulum and
Aß40 if the cleavage is in the trans-Golgi network [NATURE MEDICINE
3(9):1016-1020 (1997)]. Gamma-secretase enzyme not only produces Aß, but some
essential proteins, such as Notch [JOURNAL OF NEUROCHEMISTRY; Lathia,JD; 107(6):1471-1481 (2008)].
|
|
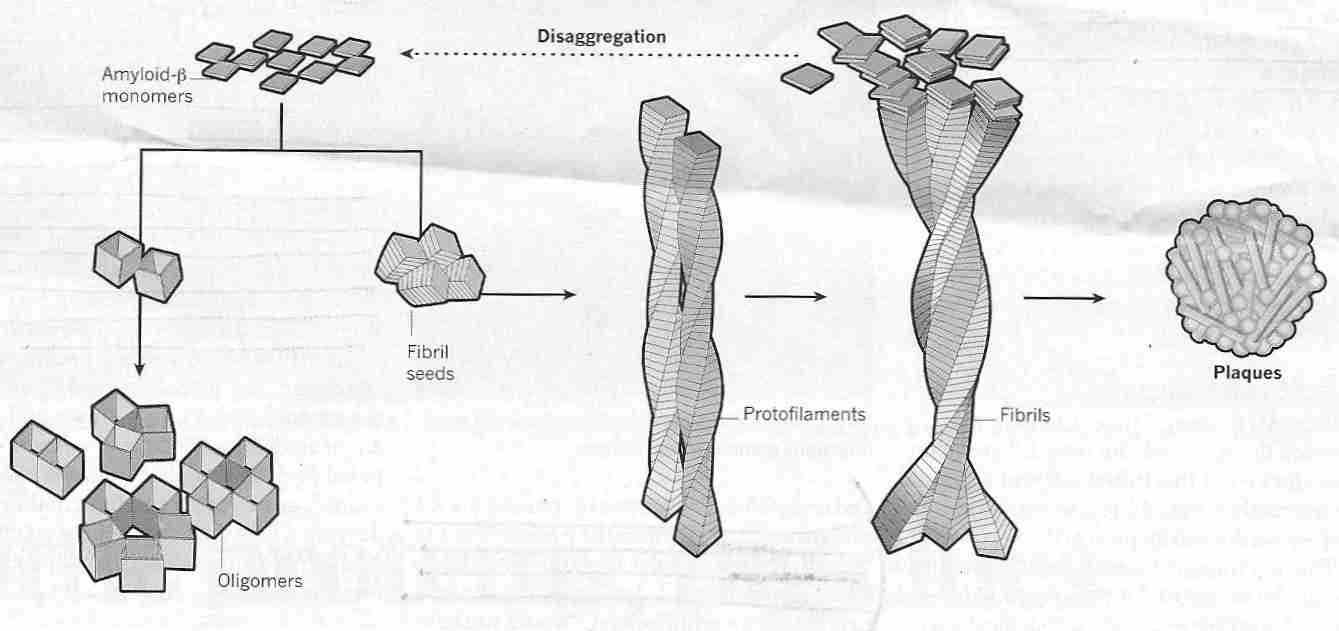
|
The 42 amino acid amyloid beta peptide (Aß42) is more hydrophobic & "sticky" (and hence aggregates more readily) than the 40 amino acid amyloid beta peptide (Aß40). Fibrils of Aß42 clump together to form amyloid plaques ("beta"[ß] refers to the "beta sheet" molecular structure of aggregated Aß). Aß40 & Aß42 are formed intracellularly, but exert damaging effects when transported outside of cells. Whereas Aß42 is the most highly concentrated amyloid-beta in neuritic plaques, Aß40 is more concentrated in cerebrovascular plaques [THE AMERICAN JOURNAL OF PATHOLOGY; Lue,L; 155(3):853-662 (1999)]. Insulin accelerates transport of intracelluar Aß to the extracellular space, which may be one reason why type-2 diabetics have a greatly increased incidence of AD [THE JOURNAL OF NEUROSCIENCE; Gasparini,L; 21(8):2561-2570 (2001)].
The most toxic form of Aß42 is believed to be in the form of soluble oligomers, i.e., strings of several Aß42 units (monomers) which are not long enough to be called polymers. There are various forms of soluble Aß42 oligomers (sometimes called ADDLs, Aß-Derived Diffusible Ligands because they can act as pathogenic ligands by attaching to synapses) and various modes of toxicity proposed for thoseoligomers [NATURE NEUROSCIENCE; Benilova,I; 15(3):349-357 (2012)]. Oligomeric Aß forms a halo around amyloid plaques that declines linearly with distance, and which is associated with excitatory synapse loss [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Koffie,RM; 106(10):4012-4017 (2009)] because Aß oligomers cause deterioration of synapses [JOURNAL OF NEUROSCIENCE; Lacor,PN; 27(4):796-807 (2007)].
Following amyloid plaque formation two processes play an important role in causing the death of neurons: (1) inflammation and oxidative damage, and (2) NeuroFibrillary Tangles (NFTs).
The two major types of brain cells that participate in the immune/inflammatory response are astrocytes and microglia. Astrocytes become more numerous in AD and these cells become activated to produce prostaglandin/arachidonic acid which mediated inflammation. Activated microglial cells produce damaging free radicals. The activities of astrocytes & microglia lead to the death of neurons.
All substances required for neuron function are manufactured in the cell body of the neurons. Neurons can be very large — with axons & dendrites extending large distances. Substances made in the cell body are transported along microtubules within neurons. Tau is an important protein that maintains the structural integrity of microtubules. But in AD the tau proteins become hyper-phosphorylated and lose the capacity to bind to microtubules. Instead, the phosphorylated tau proteins bind to each other, tying themselves in "knots" (paired helical filaments — two threads of tau wound around each other) known as NeuroFibrillary Tangles (NFTs). Neurons full of NFTs rather than functional microtubules soon die.
Injection of Aß fibrils into transgenic mice has been shown to cause NFT formation [SCIENCE; Gotz,J; 293:1491-1495 (2001)]. In transgenic mice, Aß accumulation precedes NFTs by several months [NEURON; Oddo,S; 39(3):409-421 (2003)]. Human Aß dimers (the most abundant soluble oligomers in the human brain) applied to cultures of rat hippocampal neurons have induced hyperphosphorylation of tau, thereby disrupting the microtubule cytoskeleton [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Jin,M; 108(14):5819-5824 (2011)].
With these facts in mind, the second sketch of the amyloid cascade would be:
APP ⇒ Aß42 ⇒ fibrillar Aß and oligomers ⇒ amyloid plaques
amyloid plaques ⇒ inflammation and NFTs ⇒ neuron death and synapse loss
The question of whether the central mechanism of AD neurodegeneration is beta-amyloid or NeuroFibrillary Tangles (NFTs) of tau-protein has been characterized as a "religious war" between the "tauists" and the "ßaptists". It is possible that amyloid plaques are an early event and that NFTs are a late event of an underlying process of AD that makes each event independent, but much evidence supports the view that Aß increases NFT formation.
Application of amyloid plaque to cultured neurons and injection of amyloid plaque into the brains of non-human primates both lead to NFTs. Amyloid beta may facilitate Ca2+ entry into neurons, causing calcium-activated kinases to excessively phosphorylate tau protein leading to NFTs. Some researchers have found evidence that beta-amyloid fibrils form pores in neurons leading to calcium influx and the neuron death associated with AD [NATURE 418:291 (2002)] and [CHEMICAL & ENGINEERING NEWS 80(32):31-34 (2002)]. Once amyloid plaques have become manifest, removal of those plaques is of little or no value in preventing neurodegeneration [LANCET; Holmes,C; 372:216-224 (2008)].
The definition of AD requires the existence of both tangles and plaques. Dementia due to tangles without plaques is called FrontoTemporal Dementia. A histological autopsy of the brains of deceased elderly persons showed tangles preceded plaques in one third of cases, and plaques preceded tangles in none of the cases [NEUROBIOLOGY OF AGING; Schonheit,B; 25(6):697-711 (2004)]. Commentators argued that this and related studies supports the hypothesis that Aß plaques and NFTs form independently, but where Aß is present NFT formation is accelerated [NEUROBIOLOGY OF AGING; Price,JL; 25(6):721-723 (2004)]. Depleting tau in transgenic mice blocks the cognitive impairment associated with Aß without reducing Aß levels [SCIENCE; Robertson,ED; 316:750-754 (2007)].
Alzheimer's Disease (AD) can be divided into forms that run in families (genetically inherited) [known as Familial Alzheimer's Disease (FAD)] and forms showing no clear inheritance pattern [known as Sporadic Alzheimer's Disease (SAD)]. FAD accounts for only a small portion (less than 10%) of AD. All FAD is early-onset — usually occurring between ages 30 to 60 — whereas SAD typically occurs after age 65.
All FADs can be cited as evidence of the amyloid cascade interpretation of AD causation — against the suggestion that NeuroFibrillary Tangles (NFTs), inflammation, or oxidative stress initiate AD (in FAD, at least). The gene that encodes tau-protein is located on chromosome 17 and is not associated with any FAD. In fact, at least half of FAD cases can be accounted for by the PS1 (Pre-Senilin 1) gene located on chromosome 14. PS1 is the predominant enzyme cleaving the gamma-secretase site. PS1 resides within the endoplasmic reticulum/Golgi complex. Abnormal proteins from the PS1 and PS2 genes apparently influence gamma-secretase enzyme causing more Aß42 peptide formation. The mutation on chromosome 21 (the chromosome that is present in triplicate in Down's Syndrome) is on the Amyloid Precursor Protein (APP) gene itself, resulting in abnormal APP protein that is preferentially cleaved by secretases to form more Aß42. (Down's Syndrome victims frequently develop AD if they reach age 40.)
GENE | CHROMOSOME | PROTEIN |
|---|---|---|
| Presenilin-1 | 14 | S182 |
| Presenilin-2 | 1 | STM2 |
| Amyloid Precursor Protein | 21 | APP |
| ApoLipoprotein e | 19 | APOE |
The mutations on chromosome 19 to the APOE gene are more complicated — more accurately described as a "risk factor" for SAD than as an FAD. APOE occurs in three common forms (alleles): APOE2, APOE3 & APOE4 representing in Caucasians 8%, 78% & 14% of total APOE, respectively. Although only 14% of Caucasians have one APOE4 allele and 2% will have two APOE4 alleles, 40% of AD patients will have at least one APOE4 allele. But each person has two copies of chromosome 19, which means there are 6 combinations of the 3 alleles when taken 2 at a time (e2/e2, e2/e3, e2/e4, e3/e3, e3/e4 and e4/e4). The percentages of Caucasians having at least one APOE2, APOE3 and APOE4 allele, respectively, are 15%, 94% and 25%. (Africans have higher APOE4 and Orientals have lower APOE4 concentrations.) The mean age of onset of AD for Caucasians with no APOE4 allele is 84.3, is 75.5 for one allele and 68.8 for two alleles. Nonetheless, many AD patients will have no APOE4 allele and many Caucasians with the APOE4 allele never develop AD. Because the APOE4 allele is neither a necessary nor a sufficient condition for AD, but is significantly correlated with AD, the APOE4 allele is described as a "risk factor" for Caucasians rather than as a genetic determinant. By contrast, the APOE4 allele has not been shown to be a "risk factor" for African-Americans or Hispanics (who have high risk of AD regardless of APOE4 allele) [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION 279(10):751-755 (1998)]. Moreover, APOE4 is not a risk factor for mortality (implying cardiovascular disease and dementia mortality) for anyone over age 75 [EXPERIMENTAL GERONTOLOGY; Heijmans,BT; 35(6-7):865-877 (2000)]. Although the APOE4 allele increases amyloid deposition on blood vessel walls, the APOE2 allele increases the likelihood of a lobar intracerebral hemorrhage (hemorrhage in the cerebrum rather than deep in the brain) [STROKE;Brouwers,HB;43(8):2120-2125 (2012)].
APOlipoproteins (APOs) are the protein portion of the lipoproteins (LDL,HDL,VLDL,etc) that transport cholesterol. APOlipoprotein constitutes nearly 60% of some HDL and as little as 1% of chylomicrons. APO AI is a structural component of HDL, whereas APO B100 is particularly important for LDL & VLDL. Apolipoprotein E (APOE) is a 299-amino acid glycoprotein, with three alleles: APOE2, APOE3, and APOE4. Nonhuman primates only possess the APOE4 allele, but humans have also developed the APOE2 and APOE3 alleles to facilitate cholesterol transport. The most common form, APOE3, has cysteine at position 112 and arginine at position 158. APOE2 has arginine at both those positions and APOE4 has cysteine at both those positions. Neurotoxicity due to Aß is mediated, at least in part, by the lipid peroxidation product 4-HydroxyNonEnal (HNE). The cysteine residue of APOE3 and (especially) the two cysteine residues of APOE2 protect against HNE neurotoxicity. APOE4 has no cysteine residues and is therefore not very protective against covalent modification of proteins by HNE [JOURNAL OF NEUROCHEMISTRY; Pedersen,WA; 74(4):1426-1433 (2000)].
Like the other apolipoproteins, APOlipoprotein E (APOE) is synthesized in the liver. But APOE is also independently synthesized in brain astrocytes (and, to a lesser extent in brain oligodendrocytes) — and does not cross the blood-brain barrier.
Cholesterol is an essential constituent of all cell membranes, which helps provide membrane fluidity. APOE plays a significant role in lipid/cholesterol transport by acting as a binding site for LDL (Low-Density Lipoprotein) receptors — allowing for lipids/cholesterol to be assimilated into cells. The human brain has high levels of myelin to facilitate axon conduction speed and information processing. Myelin is produced by oligodendrocytes. The brain contains 25% of the body's membrane cholesterol — and up to 80% of brain cholesterol is in myelin. Cholesterol allows for the tight packing of membranes seen in myelin sheaths. Myelin repair is dependent upon cholesterol production, recycling, and transport, which is in turn dependent upon APOE [NEUROBIOLOGY OF AGING; Bartzokis,G; 32(8):1341-1371 (2011)].
APOE mobilization of cholesterol in the Central Nervous System (CNS) is apparently of particular importance for synapse plasticity & repair of damaged neurons [JOURNAL OF NEUROCHEMISTRY 84:1215-1236 (2003)]. APOE is the major lipoprotein for lipid transport in the cerebrospinal fluid and between cells in the brain tissue itself. APOE could serve to remove oxidized lipids (including oxysterols) from the brain. APOE gene expression has been shown to decrease more than 5−fold in the cerebral cortex of mice as they age [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Jiang,CH; 98(4):1930-1934 (2001)].
The APOE4 allele is associated with higher plasma cholesterol and an even higher risk of Alzheimer's Disease (AD). Aß binds to both copper and cholesterol, fostering the oxidation of cholesterol to compounds that are extremely toxic to neurons [JOURNAL OF BIOLOGICAL CHEMISTRY; Nelson,TJ; 280(8):7377-7387 (2005)]. The APOE2 allele is associated with lower cholesterol levels and lower AD risk. Having one rather than two APOE4 alleles is a risk factor for women, but not for men. A woman with one APOE4 allele has 4 times the AD risk of a woman with no APOE4 allele. A person with two APOE4 alleles has as much as 16 times the AD risk [INTERNATIONAL JOURNAL OF CLINICAL PRACTICE 56(3):197-203 (2002)].
The APOE2 allele binds & removes amyloid-beta more avidly than APOE3, whereas APOE4 apparently does not bind amyloid-beta at all [JOURNAL OF NEUROCHEMISTRY 84:1215-1236 (2003)], except to promote amyloid ß-sheet formation [BIOCHEMISTRY 40(49):14995-15001 (2001)]. APOE3 inhibits amyloid ß-sheet formation, whereas APOE4 does not [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Evans,KC; 92(3):763-767 (1995)]. APOE4 stabilizes Aß in the toxic oligomeric form [FASEB JOURNAL; Cerf,E; 25(5):1585-1595 (2011)]. APOE2 and APOE3 have been shown to clear Aß from the brain more effectively than APOE4 [SCIENCE TRANSLATIONAL MEDICINE; Castellano,JM; 3(89):1-11 (2011)].
APOE2 protects cultured neurons most effectively from amyloid-beta generated hydrogen peroxide, APOE4 gives the least protection and APOE3 gives intermediate protection [NATURE GENETICS 14:55-61 (1996)]. Part of the anti-oxidant superiority of APOE2 above APOE3, and APOE3 above APOE4 may be due to the ability to bind metals like copper & iron insofar as APOE4 has the least metal-binding capacity [JOURNAL OF NEUROPATHOLOGY AND EXPERIMENTAL NEUROLOGY 60(8):759-767 (2001)]. Vitamin E affords the least protection against oxidative stress to human brain vascular smooth muscle cells in culture of the APOE4 genotype [CLINICAL NEUROSCIENCE 13(4):465-468 (2002)].
Individuals having both APOE4 alleles have been shown to have a smaller hippocampus. A study of cognitively normal persons aged 50-63 having both APOE4 alleles showed a 25% decline in cerebral metabolic rate over an interval of 2 years [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Reiman,EM; 98(6):3334-3339 (2001)].
APOE is normally secreted by astrocytes following neuronal damage. Cerebrospinal fluid Aß42 rises markedly after severe brain trauma [JOURNAL OF NEUROCHEMISTRY 71:2505-2509 (1998)]. Fatality following severe head injury is most strongly associated with the APOE4 genotype [NEUROPATHOLOGY AND APPLIED NEUROBIOLOGY 26:124-132 (2000)]. Head injury with loss of consciousness increases the odds of getting AD by nearly 10 times, and head injury without loss of consciousness increases the odds by more than 3. The odds of AD for those with both APOE4 genes is about 3 times as great with head injury and 8 times as great without head injury by comparison to those lacking the APOE4 gene [NEUROLOGY 54:1316-1323 (2000)]. Vascular damage, inflammation and APP mobilization have been suggested as reasons for the correlation, but APOE synthesis normally increases several hundred-fold following nervous system injury. Even without head injury, APOE4 can invoke an inflammatory response that breaches the blood-brain-barrier [NATURE; Bell,RD; 485:512-516 (2012)].
High levels of cholesterol are associated with increased risk of AD. Rabbits fed cholesterol showed twice the beta-amyloid in the hippocampal cortex as controls [ANNALS NEW YORK ACADEMY OF SCIENCES 903:335-345 (2000)]. Cholesterol binds avidly to aggregated amyloid-beta, reducing clearance and contributing to amyloid plaque [ANNALS NEW YORK ACADEMY OF SCIENCES 977:367-375 (2002)]. Atherosclerosis and AD are both particularly prevelant for the APOE4 genotype [LANCET 349:151-154 (1997)].
Apparently, the subcellular distribution of cholesterol affects amyloid-beta production. Beta-secretase activity occurs in the trans-Golgi network & late endosome, whereas gamma-secretase release of beta-amyloid is thought to occur in the endoplasmic reticulum [NEUROREPORT 10:1699-1705 (1999)]. Both beta-secretase & gamma-secretase activity increase with elevated cholesterol, whereas the reverse effect is seen with alpha-secretase (which is more active with lower cholesterol) [NEUROBIOLOGY OF DISEASE 9:11-23 (2002)]. Lowered cholesterol can not only reduce Aß production [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Wolozin,B; 98(10):5371-5373 (2001)], but experiments on hippocampal neurons have shown that a 70% reduction in cellular cholesterol was enough to eliminate Aß formation entirely [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Simons,M; 94(11):6460-6464 (1998)]. APOE4 is associated with increased deposition of Aß, but APOE4 has no effect on the rate of neurofibrillary tangle accumulation [ANNALS OF NEUROLOGY; Gomez-Isla,T; 41(1):17-24 (1997)].
Cholesterol deposition on cerebral vessel walls can lead to ischemia. APOE4 genotype nearly triples the chance of developing cerebral amyloid angiopathy (depositing of amyloid-beta on blood vessel walls) — which can also lead to ischemia [STROKE; Greenberg,SM ; 27(8):1333-1337 (1996)]. Ischemic release of calcium from the endoplasmic reticulum and disturbance of endoplasmic reticulum PS1 could be the ultimate cause of AD [NATURE MEDICINE 3(9):1016-1020 (1997)]. Alternatively, ischemic hypoperfusion could be causing AD by the failure to eliminate amyloid-beta from the brain [ANNALS NEW YORK ACADEMY OF SCIENCES 977:162-168(2002)]. Aß40 peptide has been shown to enhance cerebrovascular vasoconstriction and hypoperfusion [ANNALS NEW YORK ACADEMY OF SCIENCES 826:35-46 (1997)]. An abnormal drop in endothelial nitric oxide synthetase could promote inflammatory action by amyloid-beta [BRAIN RESEARCH REVIEWS 34:119-136 (2000)]. In any of these scenarios, AD is ultimately due to vascular disease.
Tau is a protein that stabilizes the skeletal scaffoding of neurons, ie, a protein that stabilizes the cytoskeletal microtubules. Tau is mainly present in the axons of neurons. A single gene on human chromosome 17 results in the production of five tau protein isoforms in the adult CNS, the longest of which contains 441 residues (ie, 441 amino acids). A number of kinases can phosphorylate (add a phosphate to) tau and a few phosphatases can dephosphorylate tau. The phosphorylation state of tau affects the protein's ability to self-associate or bind microtubules. Heat shock proteins and the chaperone protein Pin1 can also affect tau properties. The pathological isoforms of tau in Pick's disease and frontotemporal dementia are distinctive from those of Alzheimer's Disease, but the pattern in Down's Syndrome (trisomy of chromosome 21) is very similar to that seen in AD [PHYSIOLOGICAL REVIEWS; Avila,J; 84(2):361-384 (2004)].
Amyloid plaques typically appear first in the association areas of the cerebral cortex, whereas NeuroFibrillary Tangles (NFTs, also called Paired Helical Filaments, PHFs) typically begin in the entorhinal cortex. NFTs develop most frequently in large pyramidal neurons with long cortical-cortical connections (allowing for influence by amyloid plaques located some distance away). NFTs are associated with cells of origin of corticocortical projections whereas amyloid plaques are associated with the termination of corticocortical projections [EUROPEAN NEUROLOGY 37:71-81 (1997)]. The neurons being killed in the greatest numbers by NFTs are (1) the large cholinergic (acetylcholine-transmitting) neurons in the basal nucleus of Meynart (2) the large pyramidal neurons in the entorhinal cortex forwarding inputs from association cortices to the hippocampus via the perforant path and (3) output neurons in the CA1 region of the hippocampus. All three classes are output neurons [SCIENCE 278:412-419 (1997)].
There is evidence that the serotonergic (serotonin-producing) neurons in the raphe nuclei of the brainstem is a site of preferential NFT formation and neuronal loss in AD [NEUROPSYCHOPHARMACOLOGY; Meltzer,MD; 71(7):751-757 (2002)]. But other evidence contradicts this claim [THE AMERICAN JOURNAL OF PSYCHIATRY; Hendricksen,M; 161(6):1096-1102 (2004)].
Amyloid beta is always a feature of AD, but NFTs are not. Amyloid plaques affect sensory & motor areas of the cerebral cortex as well as association areas, but NFTs are restricted to association areas [ANNALS OF NEUROLOGY; Gomez-Isla,T; 41(1):17-24 (1997)]. Amyloid plaque is insufficient to cause the cell death of AD whereas the presence of NFTs is always associated with AD neurodegeneration [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Rapoport,M ; 99(9):6364-6369 (2002)]. Tau knock-out mice (ie, transgenic mice with no gene for tau) are resistant to Aß-induced toxicity.
In cases of AD where NFTs are absent, cell death is almost invariably associated with the Lewy bodies of Parkinsonism dementia. Lewy bodies are similar to NFTs, but are composed of ubiquitin & phosphorylated neurofilament rather than tau. Lewy bodies are typically found in the amygdala & limbic areas of the cerebral cortex (particularly the anterior cingulate cortex) in contrast to NFTs, which are concentrated in the entorhinal cortex & hippocampus (particularly the CA1 region). On autopsy AD patients have lost most of their CA1 neurons, whereas normal elderly don't show CA1 neuron loss on autopsy [EXPERIMENTAL GERONTOLOGY 38:95-99 (2003)]. Neuron death is due to NFTs or Lewy bodies — the role of amyloid plaque is at most indirect.
Hyperphosphorylation of tau is critical for the formation of NFTs. Fibrillar Aß can induce Mitogen-Activated Protein Kinase (MAPK) leading to tau phosphorylation and hence to NFTs [JOURNAL OF NEUROCHEMISTRY 74:125-133 (2000)], although other kinases may be involved as well [JOURNAL OF MOLECULAR NEUROSCIENCE 19:249-250 (2002)]. (MAPK activity — which is crucial to T-cell activation — normally declines with aging of the immune system, but MAPK pathways are aberrantly increased in AD.) Calcium influx into neurons leading to calpain-induced hyperphosphorylation of tau also contributes to NFT formation [THE AMERICAN JOURNAL OF PATHOLOGY; Veeranna; 165(3):795-805 (2004)]. But inhibition of protein phosphatases, particularly PP2A, is reportedly more responsible for hyperphosphorylated tau than kinases [JOURNAL OF BIOLOGICAL CHEMISTRY; Planel,E; 276(36):34298-34306 (2001)]. Pin1 enzyme both protects against excessive Aß production and excessive tau phosphorylation, so damage to Pin1 could lead to AD [NATURE; Pastorino,L; 440:528-533 (2006)].
Although Aß-antibody treatment can result in tau NFT clearance in the early stages of AD, the same cannot be said for later stages. Not only do NFTs become glycated & oxidized, but they bind to proteasomes, inhibiting the ability of those organelles to remove damaged proteins from cells [JOURNAL OF NEUROCHEMISTRY; Keck,S; 85(1):115-122 (2005)]. Proteasome inhibition can lead to increased Aß formation in a vicious cycle of AD [BIOCHEMICAL JOURNAL; Flood,F; 385(Pt 2):545-550 (2005)]. Moreover, ubiquitin-dependent degradation of protein is inhibited by Aß and ubiquitin is concentrated in NFTs [JOURNAL OF BIOLOGICAL CHEMISTRY; Gregori,L ; 270(34):19702-19708 (1995)].
The toxicity of Aß42 (amyloid-beta) is often attributed to the aggregation of this peptide into a ß-sheet structure of ordered fibrils [BIOCHEMICAL JOURNAL 311(Pt 1):1-16 (1995)]. Acidic conditions (such as exist in lysosomes and inflammation) enhance amyloid-beta aggregation. Cross-linking with Advanced Glycation End-products (AGEs) stabilizes amyloid plaques and accelerates the formation of ß-sheets. The Receptor for Advanced Glycation End-products (RAGE) may mediate the activation of microglia potentiation of a positive feedback loop of immune/inflammatory activation [NATURE 382:685-691 (1996)].
During development, RAGE is a cellular receptor for amphoterin, a protein that mediates neurite outgrowth. But RAGE can act as a receptor for both AGEs and Aß. Activation of RAGEs by Aß and AGEs results in the expression of more RAGEs, a positive feedback-loop that contributes to Aß toxicity [CIRCULATION RESEARCH; Schmidt,AM; 84(5):489-497 (1999)]. Aß interaction with RAGEs on endothelial cells leads to Aß transport across the blood brain barrier (BBB) as well as the expression of pro-inflammatory cytokines in those cells [NATURE MEDICINE; Deane,R; 9(7):907-913 (2003)].
Neurons in the brain are supported & nurtured by glial cells, classified as either macroglia (astrocytes & oligodendrocytes) or microglia. The microglia of the brain serve a similar function as macrophages outside of the brain. Normally the microglia are few in number, but they multiply in response to injury & infection and are concentrated around amyloid plaques — along with a subset of astrocytes (reactive astrocytes). Amyloid-beta activation of microglia causes them to produce inflammatory cytokines like InterLeukin-1ß (IL-1ß) & Tumor Necrosis Factor alpha (TNF−α). Amyloid-beta also activates the transcription factor NF−κB which increases cytokine production by neurons as well as by microglia [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Kaltschmidt, B; 94(6):2642-2647 (1997) and PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Akama,KT; 95(10):5795-5800 (1998)]. Microglia induce enzymes such as nitric oxide synthetase — generating nitric oxide leading to peroxynitrite and oxidative stress.
IL-1ß further aggravates the immune/inflammatory response by promoting more APP synthesis and by promoting the production of more Aß-binding proteins by astrocytes [NEUROBIOLOGY OF AGING 22:885-893 (2001)]. Over-expression of interleukin-1 near amyloid plaques may promote the phosphorylation of tau protein, leading to the formation of NeuroFibrillary Tangles (NFTs) and neuron death [NEUROCHEMISTRY INTERNATIONAL 39:341-348 (2001)].
The idea that a positive feedback-loop of immune/inflammatory response is central to the damaging processes of Alzheimer's Disease (AD) is reinforced by the fact that epidemiological studies have shown significant reductions in the rate of AD among people taking anti-inflammatory drugs for conditions unrelated to AD. The relative protection from AD seen from the anti-inflammatory drugs is in excess of 50% for persons taking the drugs for more than two years [NEUROBIOLOGY OF AGING 22:799-809 (2001)]. The effect is seen for Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) such as ibuprofen & indomethocin rather than for glucocorticosteroids, which are normally seen as having more profound anti-inflammatory effect.
More than a hundred years ago innoculation of aluminum phosphate into rabbit brain was demonstrated to produce NeuroFibrillary Tangles (NFTs) resembling the NFTs of Alzheimer's Disease (AD). Numerous epidemiological studies have indicated a correlation of aluminum in drinking water with the prevalence of AD, whereas studies of aluminum occupational exposure and aluminum in antacids have shown no correlation [BRAIN RESEARCH BULLETIN 55(2):187-196 (2001)]. Aluminum concentration is elevated in NFTs & amyloid plaques, but this may be an effect of AD rather than a cause. Aß42 may induce lipid peroxidation in the absence of metal catalysts [NEUROBIOLOGY OF AGING; Butterfield,DA; 23(5):655-664 (2002)]. Aß42 enhances superoxide production by macrophages [JOURNAL OF NEUROSCIENCE RESEARCH; Klegeris,A; 49(2):229-235 (1997)].
Mercury is also elevated in the AD brain. Mercury can bind to tubulin — the primary protein constituent of microtubules — thereby interfering with microtubule assembly. Zinc & selenium may protect against mercury neurotoxicity.
Although the role of aluminum remains controversial, recent research has produced more definitive information about zinc, copper & iron — all of which are enriched in amyloid-beta plaques in AD. All three metals lead to Aß aggregation, but chelation can completely reverse metal-induced precipitation of Aß [NEURON; Cherny,RA; 30(3):665-676 (2001)]. Copper particularly mediates Aß toxicity, whereas zinc inhibits toxicity [JOURNAL OF BIOLOGICAL CHEMISTRY; Maynard,CJ; 277(47):44670-44676 (2002)]. Aß is not toxic to neurons in the absence of Cu2+ [JOURNAL OF BIOLOGICAL CHEMISTRY; Opazo,C; 277(43):20302-40308 (2002)]. Aß converts Cu2+ and Fe3+ to Cu+ and Fe2+, both of which generate free radicals by the Fenton Reaction [JOURNAL OF BIOLOGICAL CHEMISTRY; Cuajungco,MP; 275(26):19439-19442 (2000)]. Fe3+ induces aggregation of phosphorylated NFTs, but this aggregation can be reversed by reducing Fe3+ to Fe2+ [JOURNAL OF NEUROCHEMISTRY; Yamamoto,A; 82(5):1137-1147 (2002)]. The copper & iron in these plaques generate hydrogen peroxide leading to oxidative damage [BIOCHEMISTRY 38(24):7609-7616 (1999)]. Although zinc binds-to and precipitates amyloid-beta, it may have a protective effect by displacing copper & iron enriched in amyloid-beta plaques in AD. On the other hand, there is evidence that zinc can initiate plaque formation by its ability to bind to Aß under non-acidic conditions and by creating the inflammation which leads to acidity. Under acidic conditions — such as exists in inflammed tissue — copper displaces zinc [BRAIN RESEARCH REVIEWS 41:44-56 (2003)]. Copper enhances ß-sheet formation of amyloid-beta fibrils and this enhancement is potentiated by APOE4 [BIOCHEMISTRY 38(14):4595-4603 (1999)]. Membranes containing oxidatively damaged phospholipids also promote amyloid ß-sheet formation [BIOCHEMISTRY 39(32):10011-10016 (2000)].
In its free state amyloid-beta has antioxidant properties which have beneficial effects for neurons. But amyloid-beta aggregation by acidic conditions and by copper, iron, zinc & aluminum results in the highly toxic & pro-oxidant ß-sheets. The binding is particularly strong for copper. Copper binds more strongly to Aß42 than to Aß40 and copper is a greater catalyst of free radical formation than are the other metals [FREE RADICAL BIOLOGY & MEDICINE 31(9):1120-1121 (2001)]. Cu2+ bound to free Aß42 is reduced by O2 to produce H2O2 [JOURNAL OF BIOLOGICAL CHEMISTRY; Opazo,C ; 277(43):40302-40308 (2002)]. Thus, copper, and to a lesser extent iron, appear to be the most serious metal toxicities in AD.
Polymerization of ß-amyloid peptide may be significantly accelerated by the presence of Advanced Glycation End-products (AGEs) [BRAIN RESEARCH REVIEWS 23:134-143 (1997)]. AGEs are formed by glycation of proteins by reducing sugars, followed by oxidation.
The most prominent marker of DNA free radical damage is 8-hydroxydeoxyguanosine (8-OHdG, equivalent to 8-oxo-7,8-dihydroguanine, 8-oxoG). Levels of 8-OHdG are 18 times higher than normal in intact DNA from the cerebrospinal fluid of Alzheimer's Disease patients [ARCHIVES OF NEUROLOGY 58:392-396 (2001)]. Severity of cognitive impairment & neurodegeneration correlates with measures of lipid peroxidation in the urine, blood and cerebrospinal fluid of AD patients [ANNALS OF NEUROLOGY 48(5):809-812 (2000)]. Oxidative damage to mitochondrial DNA reportedly precedes NFT formation [JOURNAL OF NEUROSCIENCE; Hirai,K; 21(9):3017-3023 (2001)].
Oxidized enzyme products of arachidonic acid & DHA are significantly higher in the cerebrospinal fluid of AD patients compared to age-matched controls. Arachidonic acid is evenly distributed in gray & white matter, whereas DHA is particularly enriched in gray matter. Lipid peroxidation products of arachidonic acid and DHA are prominent in the cerebral cortex & hippocampus of AD patients [THE AMERICAN JOURNAL OF PATHOLOGY; Reich,EE; 158(1):293-297 (2001)]. (For details on oxidized enzyme products of arachidonic acid see Essential Fatty Acids in Cell Membranes.)
Greater insulin resistance with aging leads to higher plasma insulin which stimulates nitric oxide synthetase. Nitric oxide in mitochondria combines with superoxide to produce peroxynitrite. Peroxynitrite causes tyrosine nitration — AD patients show elevated tyrosine nitration in both neurons & glia of brain areas exhibiting AD pathology. Peroxynitrite also results from elevated nitric oxide synthetase induced by inflammation & excitotoxicity, which may explain why AD is higher in stroke & head-injury victims [THE JOURNAL OF NEUROSCIENCE; Smith,MA; 17(8):2653-2657 (1997)]. Peroxynitrite increases proton leak across the inner mitochondrial membrane, inhibits mitochondrial enzymes and inhibits antioxidant enzymes, increasing free-radical damage [THE INTERNATIONAL JOURNAL OF BIOCHEMISTRY & CELL BIOLOGY 34:1340-1354 (2002)]. Impaired mitochondrial ATP synthesis (loss of energy) activates a protein kinase that can phosphorylate tau protein leading to NFTs [GERONTOLOGY 45:289-297 (1999)]. Glutathione can protect synapses from damage by peroxynitrite [JOURNAL OF NEUROSCIENCE RESEARCH 68:776-784 (2002)].
Tau-protein is high in lysine, which would make tau highly susceptible to glycation [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Yan,S; 91(16):7787-7791 (1994)]. Indeed, Advanced Glycation End-products (AGEs) are intimately associated with NFTs and the resultant free radical generation undoubtedly makes a significant contribution to neurodegeneration [JOURNAL OF NEUROPATHOLOGY AND EXPERIMENTAL NEUROLOGY 57(10):905-914 (1998)]. In fact, it has been suggested that protein cross-linking associated with AGEs is more critical to NFT formation than phosphorylation [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Smith,MA ; 91(12):5710-5714 (1994)].
Although Aß42 is most highly concentrated in neuritic plaques, Aß40 in the soluble state may be the most toxic form of amyloid-beta [THE AMERICAN JOURNAL OF PATHOLOGY; Lue,L; 155(1):853-862 (1999)]. Nonetheless, both forms of amyloid-beta have been shown to cause neurotoxic oxidative stress [JOURNAL OF STRUCTURAL BIOLOGY 130:184-208 (2000)].
The amyloid cascade hypothesis of AD has been challenged as being a self-fulfilling prophecy insofar as detection of amyloid plaque upon autopsy is part of the definition of Alzheimer's Disease. The sponge-like absorption of metal ions by amyloid-beta may be part of an anti-oxidant defense. Lipid peroxidation precedes Aß formation in transgenic mice [THE JOURNAL OF NEUROSCIENCE; Pratico,D; 21(12):4183-4187 (2001)] and in cases of mild cognitive impairment [ARCHIVES OF NEUROLOGY; Pratico,D; 59(6):972-976 (2002)] which frequently preceeds AD [STROKE; Meyer,JS; 33(8):1981-1985 (2002)]. Oxidative damage is actually reduced in association with both Aß & NFTs, and there is evidence that both are associated with antioxidant defense [JOURNAL OF NEUROPATHOLOGY AND EXPERIMENTAL NEUROLOGY; Nunomura,A; 60(8):759-767 (2001)]. In Down's Syndome patients (who nearly always develop AD) markers of oxidative stress are evident in neurons decades before Aß accumulation [JOURNAL OF NEUROPATHOLOGY AND EXPERIMENTAL NEUROLOGY; Nunomura,A; 59(11):1011-1017 (2000)].
Neurons are normally non-dividing (post-mitotic) cells, but neurons that have entered an aberrant cell cycle are frequently found in Alzheimer's Disease (AD). Aberrant cell cycle induction may be the primary cause of neuron death in AD, and precede Aß as well as NFT formation. Counts of hippocampal neurons in both AD and mild cognitive impairment patients show that 5-10% of neurons have cell cycle markers [THE JOURNAL OF NEUROSCIENCE; Yang,Y; 23(7):2557-2563 (2003)] suggesting that cell cycle antigens could be of benefit in early detection of AD.
Amyloid-beta toxicity may be mediated, at least in part, by the ability of that protein to induce mitosis. Aß is not toxic to glial cells, which normally proliferate, but cell cycle induction of neurons by Aß leads to cell death [THE FASEB JOURNAL; 13(15):2225-2234 (1999)]. Cells die in either an uncontrolled manner (necrosis) or by an enzyme-engineered suicide known as apoptosis. The enzymes of apoptosis are CASPases — Cysteine-dependent ASPartate-specific proteases. The ringleader of the caspases is the inflammatory cytokine InterLeukin-1ß (IL−1ß, also known as caspase−1). Although amyloid-beta seems to initiate apoptosis, there is little evidence that the process runs to completion or that simple apoptosis is a significant contributor to neuron death in AD [JOURNAL OF NEUROPATHOLOGY AND EXPERIMENTAL NEUROLOGY 60(9):829-838 (2001)]. But caspases can nonetheless contribute significantly to Aß pathology.
The fact that clearance of Aß from hippocampal tissue by injection of anti-Aß antibody leads to removal of early tau pathology provides strong evidence for the amyloid-cascade hypothesis [TRENDS IN MOLECULAR MEDICINE; LaFerla,FM; 11(4):171-176 (2005)]. Aß toxicity is considerably mediated through caspase−12 enzyme in the endoplasmic reticulum [NATURE; Nakagawa,T; 403:98-103 (2000)]. Caspase-cleaved tau protein accelerates the aggregation and filament formation of full-length tau, which co-localizes with Aß, aggrevating the tangles [THE JOURNAL OF CLINICAL INVESTIGATION; Rissman,RA; 114(1):121-130 (2003)]. As a caspase substrate, tau is cleaved at a single site: Asp−421/Ser−422 [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Gamblin,TC; 100(17):10032-10037 (2003)].
The fact that the genetic mutations affecting Aß production (PS1, PS2 and APP proteins) are behind Familial Alzheimer's Disease (FAD, inherited AD) has been taken as strong evidence for the Amyloid Cascade Hypothesis. But mutations in these proteins can also be used as evidence for an "Aberrant Cell Cycle Hypothesis" of AD. Overexpression of APP can drive neurons into a cell cycle leading to apoptosis [JOURNAL OF BIOLOGICAL CHEMISTRY; Chen,Y; 275(12):8929-8935 (2000)]. PS2 overexpression leads to cell cycle arrest preceding apoptosis in cell cultures [AMERICAN JOURNAL OF PATHOLOGY; Janicki,SM; 155(1):135-144 (1999)]. Mice genetically disposed to hyperphosphorylated, aggregated tau will display neurodegeneration associated with abnormal neuronal cell-cycle re-entry leading to cell death [JOURNAL OF NEUROSCIENCE; Andorfer,C; 25(22):5446-5454 (2005)]. Three variants of PS1 mutations lead to a degree of cell cycle inhibition that correspond to the age of onset of FAD in humans afflicted with those mutations [NEUROBIOLOGY OF AGING; Janicki,SM; 21(6):829-836 (2000)].
A "Two Hit Hypothesis" of AD suggests that chronic mild oxidative stress and extemely slow apoptosis associated with aberrant cell cycles in neurons are both necessary conditions for Alzheimer's Disease, but that neither alone is a sufficient cause [THE LANCET NEUROLOGY; Zhu,X; 3:219-226 (2004) and Alzforum: Current Hypothesis]. In this model, Aß & NFTs are secondary to the destructive processes leading to AD.
Several early epidemiological studies claimed that cigarette smoking has a protective effect against Alzheimer's Disease [NEUROEPIDEMIOLOGY 13:131-144 (1992)]. Such studies have been criticized on many methodological grounds, including survival bias & recall bias [ADDICTION 97:15-28 (2002) and BIOLOGICAL PSYCHIATRY 49:194-199 (2001)]. More nicotine receptors and fewer senile plaques are not surprising among autopsies of smokers if they are dying at a younger age than non-smokers.
More recent prospective (cohort) studies found opposite results from the earlier cross-sectional or prevalence studies. A prospective Rotterdam Study found that the incidence of AD is more than double for smokers as compared to non-smokers (smoking was not an additional risk factor for those having the APOE4 allele) [THE LANCET 351:1840-1843 (1998)]. The Honolulu Heart Program (a longitudinal cohort study) also found more than twice the risk for AD among medium & heavy smokers as compared to non-smokers [NEUROBIOLOGY OF AGING 24:589-596 (2003)]. Large scale observational studies show nearly twice the risk of AD for smokers, as compared to non-smokers or (to a lesser extent) former smokers [AMERICAN JOURNAL OF EPIDEMIOLOGY; Anstey,KJ; 166(4):367-378 (2007)].
M2 muscarinic receptors and nicotinic receptors are markedly decreased in AD. Nicotine administration to transgenic mice has significantly reduced Aß42 plaques as compared to sucrose-administered controls [JOURNAL OF NEUROCHEMISTRY 81:655-658 (2002)]. Nicotine administration into rat hippocampus produces lasting elevation of Nerve Growth Factor (NGF), enhancing acetycholine production & release [BIOLOGICAL PSYCHIATRY 49:185-193 (2001)]. Studies of AD patients have shown beneficial effects of nicotine administration [BIOLOGICAL PSYCHIATRY 49:200-210 (2001)]. Nonetheless, a mouse model has shown a worsening of tau protein pathology (NFTs) due to nicotine administration [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Oddo,S; 102(8):3046-3051 (2005)].
Tobacco smoke is a lethal substance, so claims of its possible benefits against AD should be viewed with caution. In a 40-year longitudinal study of tens of thousands of British Physicians, the death rate between the ages of 35 to 69 was only 20% for non-smokers as compared to 41% for smokers as a whole and 50% for those who smoked more than 25 cigarettes per day. Of those who survived to age 70, nonsmokers had twice the chance of living to age 85 as smokers [BRITISH JOURNAL OF MEDICINE 309:901-911 (1994)]. Even if nicotine is proven decisively to be of benefit in AD, it should be administered as nicotine patches rather than as tobacco smoke — which contains carbon monoxide, cadmium and thousands of other toxins which may contribute to AD. If AD is a vascular disease, the damaging effects of the toxins in tobacco smoke on the vasculature alone could easily outweigh possible increases in brain NGF. And if AD is not a vascular disease, tobacco smoke certainly nonetheless contributes to vascular dementia.
Intracellular accumulation of Aß may be at least as significant a contributer to Alzheimer's Disease (AD) as extracellular accumulation. There is evidence that co-localization of intracellular chaperone proteins with Aß contributes to Aß metabolism and toxicity [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Fonte,V; 99(14):9439-9444 (2002)].
Some investigators suggest that the degenerative changes of AD are preceded by vascular or atherosclerotic changes that reduce blood flow and thereby induce ischemic & oxidative stresses leading to AD [STROKE; de la Torre,JC; 33(4):1152-1162 (2002)]. Transgenic mice that overexpress APP show that amyloid-beta affects cerebrovascular regulation — reducing blood flow. There is also evidence that cleavage of amyloid-beta from APP is enhanced by ischemia [STROKE; Iadecola,C; 34(2):335-337 (2003)].
The relative risk of developing AD is reportedly 12-fold greater for individuals positive for Herpes Simplex Virus Type 1 (HSV1) and the APOE4 allele than for persons with one or neither of these factors [HERPES; Itzhaki,R; 11(suppl 2):77A-82A (2004)]. Infected neurons show a rapid decline of full length APP and an increase in an APP fragment containing the Aß sequence [BMC MICROBIOLOGY; Shipley,S; 5(48):(2005)]. Cultured neuronal and glial cells infected with HSV1 show increased Aß42 deposits [NEUROSCIENCE LETTERS; Wozniak,WA; 429(2-3):95-100 (2007)]. The great majority of amyloid plaques contain HSV1, especially in AD patients, suggesting that compromised immune systems in the elderly increase HSV1 infection, leading to chronic inflammation and AD [JOURNAL OF PATHOLOGY; Wozniak,MA; 217(1):131-138 (2009)].
Much higher levels of mitrochondrial DNA Control Region (CR) mutations have been found in AD patients compared to age-matched controls. These mutations could be an effect or cause of AD, or both [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Coskun,PE; 101(29):10726-10731 (2004)].
Early detection of AD pathology can allow for early treatment and the greatest slowing of the progression of the disease. The benefit of early detection will be especially great if means are found to halt or reverse the degenerative processes of AD.
A screen of the serum of AD patients for potentially diagnostic Immunoglobulin G (IgG) antibodies has yielded promising candidates [CELL; Reddy,MM; 144(1):132-142 (2011)]. Because oxidative damage is a notable part of AD neurodegeneration, such damage has been suggested as a possible biomarker [AGEING RESEARCH REVIEWS; Mangialasche,F; 8(4):285-305 (2009)].
Autopsy analysis of the CerebroSpinal fluid (CSF) of AD patients indicates reduced levels of Aß and increased concentrations of tau proteins [ANNALS OF NEUROLOGY; Shaw,LM; 65(4):403-413 (2009)]. The same result has been found in living AD patients [ARCHIVES OF NEUROLOGY; Tapiola,T; 66(3):382-389 (2009)]. Aß accumulation in insoluble plaques results in low levels of CSF Aß [ANNALS OF NEUROLOGY; Fagan,AM; 59(3):512-519 (2006)]. Analysis of the CSF of elderly individuals confirm that a decline in CSF Aß precedes the elevation in CSF tau protein [ARCHIVES OF NEUROLOGY; De Meyer,G; 67(8):949-956 (2010)]. A 2-year study of 7 patients with mild cognitive impairment showed increasing CSF hyperphosphorylated tau and decreasing CSF Aß42 [NEUROBIOLOGY OF AGING; de Leon,MJ; 27(3):394-401 (2006)]. Although CSF Aß is a good biomarker of AD progression, plasma Aß is not — and is unrelated to CSF Aß [NEUROBIOLOGY OF AGING; Hansson,O; 31(3):357-367 (2010)]. Decreased CSF Aß is not definitive for AD, however, because CSF Aß decreases in other forms of dementia [NEUROBIOLOGY OF AGING; Craig-Schapiro,R; 35(2):128-140 (2009)].
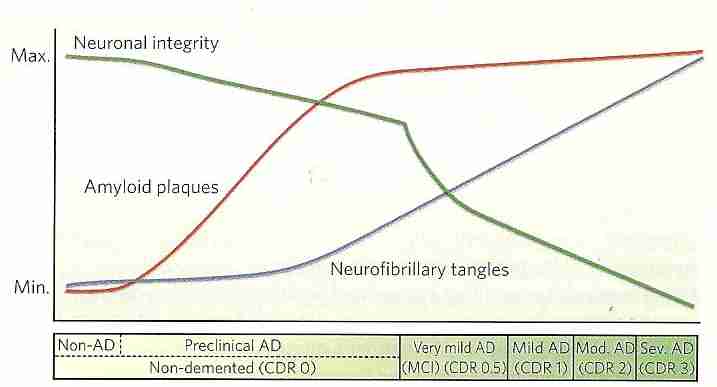
|
PiB (Pittsburg compound B) binds to fibrillar Aß (but not soluble Aß or diffuse plaques), allowing for Positron Emission Tomography (PET) before behavioral symptoms of AD are seen [BRAIN; Knight,WD; 134(Pt 1):293-300 (2011)]. But PiB has a half-life of 20 minutes, requiring an on-site cyclotron.
Florbetapir F 18 (AMYViD) is a radioactive compound with a that binds to Aß, has a longer half-life than PiB, and is well visualized by PET. A strong correlation has been seen between Florbetapir PET imaging and Aß deposition in a post-mortem study of elderly brains [ALZHEIMER DISEASE AND ASSOCIATED DISORDERS; Choi,R; 26(1):8-16 (2012)]. PET visualization of 10 subjects with AD and 10 young, healthy controls identified all the AD patients as Aß-positive, and all of the controls as Aß-negative [JOURNAL OF NUCLEAR MEDICINE; Joshi,AD; 53(3):378-384 (2012)].
Magnetic resonance imaging (MRI) of cerebral cortex thickness of elderly subjects showed that characteristic structural changes can be identified two years before diagnostic behavioral symptoms become manifest [BRAIN; Querbes,O; 132(Pt 8):2036-2047 (2009)]. MRI abnormalities appear with behavioral symptoms and the neurodegeneration indicated with MRI occurs in parallel with cognitive decline [LANCET NEUROLOGY; Jack,CR; 9(1):119-128 (2006)]. An MRI study of about 500 subjects for six months showed that in AD subjects ventricular enlargement was 60% greater than subjects with mild cognitive impairment, and four times greater than normal elderly controls — and ventricular enlargement was greater in AD victims having one APOE4 allele than those having none [BRAIN; Nestor,SM; 131(9):2443-2454 (2008)].
Insofar as amyloid plaque accumulation maximizes before even milder symptoms of AD appear, to be useful diagnostic predictors, biomarkers for Aß would have to be screened-for routinely in healthy subjects. Once AD has developed, removal of Aß does not stop neurodegeneration [LANCET; Holmes,C; 372:216-224 (2008)], but early removal of Aß may prevent AD [SCIENCE TRANSLATIONAL MEDICINE; Castellano,JM; 3(89):1-11 (2011)]. PET is expensive and is not generally available in hospitals. Lumbar punctures to obtain CSF is invasive, risk infection, and could damage the spinal cord.
In the 1970s it was widely believed that Alzheimer's Disease (AD) is a disturbance of the acetylcholine functions in the brain. AD is associated with decreased density of nicotinic acetylcholine receptors in the cerebral cortex. Muscarinic acetylcholine receptors play an important role in learning & memory. Over three-quarters of cholinergic neurons in the basal forebrain nuclei are seen to be destroyed in a typical AD autopsy. Loss of choline acetyltransferase in the nucleus basalis was the first biomarker of AD. The large pyramidal neurons of the cortex & forebrain acetylcholine neurons — which are important for cognition — have more microtubules than other neurons, and it is these large neurons that show the most NeuroFibrillary Tangles (NFTs) in AD.
Feeding lecithin (phosphatidylcholine) to animals increases brain acetylcholine levels, but clinical trials using lecithin on AD patients has shown little or no improvement. Likewise, attempts to treat AD patients with drugs that stimulate acetylcholine receptors have been a failure.
Most of the FDA-approved drugs for AD treatment are inhibitors of the enzyme that breaks down acetylcholine in the synapse — acetylcholinesterase (AChE). Those drugs have been presumed to work by prolonging the lifetime of released acetylcholine. Liver toxicity and other side effects have reduced the use of tacrine. Rivastigmine, donezepil & galantamine seem comparable in their efficacy, tolerability & side-effects [BRITISH JOURNAL OF PSYCHIATRY 180:135-139 (2002)]. Rivastigmine & galantamine inhibit butyrylcholinesterase as well as AChE. Galantamine has a direct stimulant action upon nicotinic receptors. Nausea, vomiting & diarrhea are the most common side effects of the AChE inhibitors. Approximately 50% of AD patients treated with AChE inhibitor show lack of cognitive deterioration for up to two years.
There is increasing evidence, however, that the primary action of AChE inhibitors may not be primarily due to prolongation of action of acetylcholine. AChE promotes aggregation of amyloid-beta [MOLECULAR AND CELLULAR NEUROSCIENCES; Pera,M; 40(2):217-224 (2009)]. A mixture of amyloid-beta & AChE shows 3-times more aggregation of amyloid-beta than amyloid-beta alone. Glycation of AChE is increased by amyloid-beta [MECHANISMS OF AGING AND DEVELOPMENT 122:1961-1969 (2001)]. AChE inhibitors increase the amount of APP that is cleaved by alpha-secretase rather than beta-secretase [NEUROCHEMICAL RESEARCH 28(3/4):515-522 (2003)].
Since 1989 memantine has been used in Germany for AD, but was only approved by the FDA in 2003. Memantine blocks the neurotransmitter glutamate. Excitotoxicity due to excessive glutamate release is associated with inflammation & oxidative stress. AD patients taking memantine are able to live independently 6 months longer than they would without the drug. Memantine protects against excitotoxicity without interfering with glutamic signalling, while also increasing BDNF and exerting anti-oxidant effects. Although the benefits of memantine and AChE for slowing the progress of AD are statistically significant, the clinical improvement is marginal [ANNALS OF INTERNAL MEDICINE; Raina,P; 148(5):379-397 (2008)].
With the identification of the gene that produces it, beta-secretase has been renamed BACE (Beta-site APP-Cleaving Enzyme). Blockage of BACE is a promising therapy for AD. Blockage of gamma-secretase seems more problematic because the enzyme appears to be essential for the activity of the membrane "notch" protein that sends signals to the nucleus to direct cell development [EUROPEAN JOURNAL OF CLINICAL INVESTIGATION 32:60-68 (2002)].
There is evidence that a reduction in the ability of T-cells to clear amyloid-beta may underly the chronic inflammation of AD [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Monsonego,A; 98(18):10273-10278 (2001)]. Aß deposits in transgenic mice were eliminated by amyloid-beta vaccination, but human clinical trials of Aß vaccination were halted when 6% of the patients developed brain inflammations (autoimmune meningoencephalitis [JOURNAL OF INTERNAL MEDICINE; Morgan,D; 269(1):54-63 (2011)]. A flaw in the experiment may have been that the mice cleared the human Aß without completely clearing the mouse Aß, which differs in three amino acids [NEUROBIOLOGY OF AGING 24:391-395 (2003)]. Aß immunotherapy of Caribbean velvet monkeys has been shown to decrease brain Aß42 by 66% [THE AMERICAN JOURNAL OF PATHOLOGY; Lemere,CA; 165(1):283-297 (2004)]. A follow-up study on the human clinical trials showed that even removal of the amyloid plaques did not stop neurodegeneration [LANCET; Holmes,C; 372:216-224 (2008)]. Nonetheless, clinical trials continue with monoclonal antibodies against Aß [EXPERT REVIEW OF CLINICAL IMMUNOLOGY; Imbimbo,BP; 8(2):135-149 (2012)]. Passive immunization with tau antibodies has been shown to reduce tau pathology [JOURNAL OF NEUROCHEMISTRY; Boutajangout,A; 118(4):658-667 (2011)].
Intramuscular injections with desferroxamine — which binds the trivalent metal ions of iron & aluminum — was shown to halve the cognitive decline of AD patients over a two-year period [LANCET 337:1304-1308 (1991)]. EDTA and other chelators of zinc & copper have been shown to solubilize amyloid-beta from post-mortem AD tissues [JOURNAL OF BIOLOGICAL CHEMISTRY; Cherny,RA; 274(33):23223-23228 (1999)]. Transgenic mice treated with the antibiotic clioquinol had a 49% decrease in brain Aß deposits. Clioquinol selectively binds zinc & copper and crosses the blood-brain barrier, but has been associated with dangerous depletion of vitamin B12 [NEURON 30:665-676 (2001)]. A 12-week placebo-controlled, double-blind trial of chioquinol treatment showed a slowed cognitive decline in AD patients having severe dementia, but not in those with moderate dementia [ARCHIVES OF NEUROLOGY; Ritchie,CW; 60(12):1685-1691 (2003)].
A clioquinol analog named PBT2 has greater blood-brain permability, and more effectively strips copper and zinc from Aß without the chelation, which could remove essential metal ions [NEUROTHERAPEUTICS; Bush,AI; 5(3):421-432 (2008)]. PBT2 in a mouse model of AD showed considerable reduction in AD pathology associated with improved learning & memory [NEURON; Adlard,DA; 59(1):43-55 (2008)]. Initial clinical trials have been promising, while indicating no adverse effects [LANCET NEUROLOGY; Lannfelt,L; 7(9):779-786 (2008)].
The PPAR-gamma agonist rosiglitazone (a drug for treating type 2 diabetes) improves cognition and memory in patients with mild to moderate AD [PHARMACOGENOMICS JOURNAL; Risner,ME; 6(4):246-254 (2006)]. Rosiglitazone has been shown to increase dendritic spines in cultured neurons in a dose-dependent manner [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Brodbeck,J; 105(4):1343-1346 (2008)]. Insulin decreases APP metabolism, reducing Aß, in part by increasing Aß degredation. Insulin complelely eliminates ADDL (Aß oligomer)-induced synaptic spine deterioration [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); DeFelice,FG; 106(6):1971-1976 (2009)]. The insulin-sensitizing drug metformin up-regulates beta-secretase (BACE1), but in combination with insulin reduces Aß levels [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Chen,Y; 106(10):3907-3912 (2009)].
The protease cathepsin B reduces levels of Aß42 intracelluarly and extracellularly by proteolytic action [NEURON; Mueller-Steiner,S; 51(6):703-714 (2006)]. Neprilysin is also a major extracellular Aß-degrading enzyme in the brain [JOURNAL OF NEUROSCIENCE; Marr,RA; 23(6):1992-1996 (2003)]. Increasing production of natural Aß-degrading enzymes could be a tool for AD therapy and prevention.
In transgenic rat models of AD, gene delivery of Brain-Derived Neurotrophic Factor (BDNF) administered after disease onset reversed synaptic loss in the hippocampus and restored learning and memory [NATURE MEDICINE; Nagahara,AH; 15(3):331-337 (2009)]. Like memantine, cannabinoids can increase BDNF activity [BRITISH JOURNAL OF PHARMACOLOGY; Campbell,VA; 152(5):655-662 (2007)].
"Pathological chaperone" molecules which assist in the aggregation of Aß represent potential therapeutic targets [BMC NEUROSCIENCE; Wisniewski,T; 9(Suppl 2):S5 (2008)].
Plasma levels of the cytokine Tumor Necrosis Factor-alpha (TNF−α) increase with age and those levels were shown to predict mortality in institutionalized elderly patients. Blockage of TNF−α in a mouse model of AD reduces cognitive deficits produced by Aß [JOURNAL OF NEUROSCIENCE; Medeiros,R; 27(20):5394-5404 (2007)]. High TNF−α plasma levels are also independently associated with atherosclerosis as well as AD [JOURNALS OF GERONTOLOGY 54A(7):M357-M364 (1999)]. TNF−α suppresses growth hormone signal transduction and insulin sensitivity.
Etanercept, and antagonist of the inflammatory cytokine TNF−α, has shown effectiveness in reducing symptoms in an AD patient within hours of administration — and with effective maintenance with weekly doses [JOURNAL OF NEUROINFLAMMATION; Tobinick,EL; 5:2 (2008)].
Glycogen Synthetase Kinase-3 (GSK-3) is one of the enzymes that phosphorylates tau protein. Insulin down-regulates GSK-3 activity, whereas brain insulin resistance results in increased GSK-3 activity. Inhibition of GSK-3 by lithium has been shown to substantially reduce NFT formation in transgenic mice [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Noble,W; 102(19):6990-6995 (2005)].
Aggregated Aß peptide toxicity may be mediated in part by activating calcium-permeant AMPA receptors on neurons, resulting in Ca2+−influx which can activate calpain enzymes. DAPH (4,5−dianilinophthalimide) has been shown to disaggregate Aß fibrils and eliminate the Ca2+−influx [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Blanchard,BJ; 101(40):14326-14322 (2004)].
Inflammed microglia are believed to contribute to neuronal death as a result of NF-κB stimulated by Aß. Resveratrol has been shown to markedly reduce the NF-κB signalling in mixed cortical/glial cell cultures [JOURNAL OF BIOLOGICAL CHEMISTRY; Chen,J; 280(40):40364-40374 (2005)]. Resveratrol has also been shown to promote Aß degradation by proteosomes [JOURNAL OF BIOLOGICAL CHEMISTRY; Marambaud,P; 280(45):37377-37382 (2005)].
A review of the literature on AD treatment with ginko biloba concluded that 120 to 240 mg for 3 to 6 months is associated with a 3 % reduction in cognitive decline [ARCHIVES OF NEUROLOGY 55:1409-1415 (1998)]. A decade later a large randomized trial showed ginko biloba improved cognition in AD patients [JOURNAL OF NEUROLOGICAL SCIENCES; Napryenko,O; 283(1-2):224-229 (2009)] and a large review also showed cognitive improvement for AD patients [BMC GERIATRICS; Weinmann,S; 10:14 (2010)]. But a large 6-year, randomized, double-blind, placebo-controlled trial showed that ginko biloba was not effective in preventing AD [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION; Dekosky,S; 300(19):2253-2262 (2008)] — possibly implying that ginko biloba is an effective treatment, but is not effective for preventing AD.
A double-blind, placebo-controlled clinical trial with daily doses of 10 mg deprenyl (selegiline), 2000 IU vitamin E, or both, showed a 25% slowing of the progress of AD. There was no advantage to using both deprenyl & vitamin E [NEW ENGLAND JOURNAL OF MEDICINE 336(17):1216-1222 (1997)]. Supplementation with vitamins C & E can correct the low levels of these vitamins in the cerebrospinal fluid of AD patients [FREE RADICAL BIOLOLGY & MEDICINE 31(3):345-354 (2001)]. Nonsmokers with high levels of dietary vitamin C & E showed 82% reduced risk of AD over a 6-year period and nonsmokers showed an even greater reduction of risk with the high-vitamin diet [NUTRITION REVIEWS 61(2):69-73 (2003)]. (The predominant dietary form of Vitamin E is gamma-tocopherol, in contrast to Vitamin E from supplements, which is predominantly alpha-tocopherol.)
Trehalose inhibits the formation of both fibrillar and oligomeric aggregation of Aß40, but only inhibits formation of fibrillar aggregates of Aß42 [NEUROBIOLOGY OF DISEASE; Liu,R; 20:74-81 (2005)].
NSAIDs are more effective in preventing AD than in treating the disease. NSAIDs act by inhibition of the enzyme CycloOXygenase (COX). Microglia express the COX-1 form of the enzyme, whereas neurons express the COX-2 form. Although older NSAIDs block both COX-1 & COX-2, newer NSAIDs are targeted against COX-2, which causes pain and inflammation in joints. The beneficial effects for preventing AD are primarily attributed to inhibition of COX-1 in the microglia. Unfortunately, COX-1 inhibition is associated with gastrointestinal bleeding. It has been suggested that NSAIDs directly decrease Aß42 formation, independent of COX inhibition [NATURE 414:212-216 (2001)], but neither naproxen nor aspirin lower Aß42, yet both have protective effects against AD. NSAIDs (ibuprofen and indomethicin) inhibit cytokine activation of ß-secretase (BACE1) by stimulating PPAR-gamma [JOURNAL OF NEUROSCIENCE; Sastre,M; 23(30):9796-9804 (2003)]. APOE expression is induced by PPAR-gamma and Liver X Receptors (LXRs) in coordination with Retinoid X Receptors (RXRs), such that an RXR agonist can reduce Aß in an APOE-dependent manner [SCIENCE; Cramer,PE; 335:1503-1506 (2012)].
The complement system involves a collection of proteins including those designated C1 to C9 — which participates in an amplifying cascade of enzymes that results in large numbers of downstream complements (ie, a few C1s can result in very many C8s & C9s). Outside of the brain, complements participate in antigen-antibody reactions, but in the brain the complement protein C1b binds to amyloid-beta fibrils which are phagocytosed by microglia. If it is true that the fibrils are largely undegraded by the microglia, a "frustrated phagocytosis" could lead to a worsened condition due to heightened microglia activation [NEUROCHEMISTRY INTERNATIONAL 39:333-340 (2001)].
Two major epidemiological studies have indicated that a high level of dietary Vitamin E is associated with a lowered risk of AD. In one of the studies the benefit was seen only for persons without the APOE4 genotype, but in the second study the APOE4 genotype had no effect on the outcome. The second study also suggested that dietary Vitamin C may also lower risk of AD [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION 287:3223-3239 & 3230-3237 (2002)]. Vitamin E has been shown to reduce F2-isoprostane levels in the plasma of rats [FREE RADICAL BIOLOGY AND MEDICINE 34(7):795-799 (2003)] and to reduce Aß deposition in mice subjected to repetitive concussive brain injury [JOURNAL OF NEUROCHEMISTRY 90:758-764 (2004)]. (The predominant dietary form of Vitamin E is gamma-tocopherol, in contrast to Vitamin E from supplements, which is predominantly alpha-tocopherol.)
An observational study of over 6,000 people 65 years of age and older found that compared to people in the lowest fifth of niacin intake, those with higher intake had between 20% to 50% the risk of getting AD. This result was after adjustment for age, sex, race, education, ApoE type and intake of vitamins E & C as well as beta-carotene. The effect was specific to niacin, as opposed to the other B vitamins [JOURNAL OF NEUROLOGY, NEUROSURGERY, AND PSYCHIATRY; Morris,MC; 75(8):1093-1099 (2004)].
Diets high in both saturated fatty acids and copper are associated with increased risk of AD. Foods highest in copper content include beef liver, oysters, molluscs, almonds, and cocoa [BRITISH JOURNAL OF NUTRITION; Loef,M; 107:7-19 (2011)].
Melatonin (N-acetyl-5-methoxytrypamine) is a non-toxic neurohormone produced by the pineal gland. Melatonin secretion & synthesis decline sharply with aging and is profoundly lower in age-matched AD patients — most especially AD patients who are homozygous for APOE4. Melatonin significantly inhibits the formation of neurotoxic amyloid ß-sheets [BIOCHEMISTRY 40(49):14995-15001 (2001)], indicating a possible preventative benefit. Unlike Vitamin E, melatonin acts as an antioxidant through endogenous electron donation, which does not have the same potential for a pro-oxidant side effect [JOURNAL OF PINEAL RESEARCH 32:135-142 (2002)]. The capacity of melatonin to scavenge hydroxyl radicals is three orders of magnitude greater than Vitamin E [JOURNAL OF BIOLOGICAL CHEMISTRY; Chyan,Y; 274(31):21937-21942 (1999)]. Melatonin supplementation might be protective against AD.
Transgenic mice that develop AD, when given pomegranate juice from 6 to 12.5 months of age showed 50% less accumulation of soluble Aß40 and amyloid deposits in the hippocampus associated with improved learning ability, compared to controls [NEUROBIOLOGY OF DISEASE; Hartman,RE; 24:506-515 (2006)].
Epidemiological studies have shown that consumption of the omega-3 essential fatty acid DHA (found in fish oils, particularly salmon) is associated with reduced risk of AD [ARCHIVES OF NEUROLOGY 60:940-946 (2003)]. Aß42 accumulates in dendrites of AD patients and DHA treatment of AD-model mice protected against loss of dendrite protein [NEURON 42:633-645 (2004)]. DHA supplementation of aged Alzheimer mouse models reduced Aß plaques by nearly 50% in the hippocampus and parietal cortex [THE JOURNAL OF NEUROSCIENCE; Lim,GP; 25(12):3032-3040 (2005)]. DHA has also been shown to induce a 10-fold increase in transcription of the Aß-scavenger transthyretin [PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES (USA); Puskas,LG; 100(4):1580-1585 (2003)]. Transthyretin is a natural Aß-degrading enzyme that declines with aging [MOLECULAR NEURODEGENERATION; Li,X; 6:79 (2011)]. Nonetheless, a randomized, double-blind trial on patients with mild to moderate AD failed to demonstrate that DHA slowed cognitive decline better than placebo [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION; Quinn,JF; 304(17):1903-1911 (2010)]. Nicotine and Ginko bilboa also induce transthyretin transcription. Because DHA is so vulnerable to oxidation, increased intake of antioxidants has been recommended to accompany increased intake of DHA.
A randomized, placebo-controlled, double-blind 12-week trial of chromium picolinate (which reputedly increases insulin sensitivity) on subjects with mild cognitive impairment and early AD showed improved memory performance associated with increased brain activation, as indicated by functional MRI [NUTRITIONAL NEUROSCIENCE; Krikorian,R; 13(3):116-122 (2010)].
Low serum folic acid and elevated plasma homocysteine are both independent risk factors for AD [AMERICAN JOURNAL OF CLINICAL NUTRITION; Ravaglia,G; 82:636-643 (2005)]. Folic acid deficiency and excess homocysteine impair DNA repair, thereby sensitizing neurons to cell death due to DNA damage [JOURNAL OF NEUROSCIENCE; Kruman,II; 22(5):1752-1762 (2002)]. Folic acid deficiency and excess homocysteine also induce beta-secretase (BACE) and PRE1, thereby increasing Aß production — an effect that can be overcome by administration of S-Adenosyl Methionine [MOLECULAR AND CELLULAR NEUROSCIENCE; Fuso,R; 28(1):195-204 (2005)].
Inflammatory conditions stimulate inducible nitric oxide synthetase to produce more nitric oxide, which can react with superoxide to form the free-radical peroxynitrite. The phytochemical curcumin (which gives curry its yellow color) is a powerful antioxidant which (unlike alpha-tocopherol) can scavenge peroxynitrite and inhibit inducible nitric oxide synthetase (thereby inhibiting COX-2) [CARCINOGENESIS; Rao,CV; 20(4):641-644 (1999)]. Moreover, curcumin not only inhibits amyloid-beta fibril and oligomer formation, but disaggregates existing plaques [JOURNAL OF BIOLOGICAL CHEMISTRY; Yang,F; 280(7):5892-5901 (2005)]. Curcumin can also lower brain levels of the pro-inflammatory cytokine IL−1ß nearly 60% in transgenic mice [THE JOURNAL OF NEUROSCIENCE; Lim,GP; 21(21):8370-8377 (2001)]. Curcumin can also increase insulin sensitivity [FOOD AND CHEMICAL TOXICOLOGY; Kang,C; 48:2366-2373 (2010)]. These facts could explain reports of significantly lower incidence of AD among the elderly in India [NEUROLOGY; Chandra,V; 57(67):985-989 (2001)].
The natural hormone DeHydroEpiAndrosterone (DHEA) increases APP synthesis & secretion — which is thought to be neuroprotective — while reducing the formation of Aß. Epidemiological studies indicate an association between AD and an age-related decline in DHEA. For these reasons DHEA supplementatioin may reduce the risk of AD [LIFE SCIENCES 59(19):1651-1657 (1996)].
Estrogen reduces the formation of amyloid-beta by increasing alpha-secretase activity. Estrogen also enhances amyloid-beta uptake by microglia. Estogen opposes neuron atrophy by stimulating nerve growth factors. Estrogen reduces the activation of the pro-inflammatory transcription factor NF-κB by amyloid-beta and enhances cholinergic projections from the basal forebrain [NEUROCHEMICAL RESEARCH 28(7):1003-1008 (2003)]. Both estrogens & androgens protect cultured human neurons from Aß toxicity by increasing levels of heat shock protein (Hsp70) [THE JOURNAL OF NEUROSCIENCE; Zhang,Y; 24(23):5315-5321 (2004)].
Estrogen replacement therapy in post-menopausal women can increase cerebral blood flow and improve cognitive performance. The brains of men maintain the ability to convert testosterone to estrogen with advancing age, which may explain the greater prevalence of AD among women [JOURNAL OF THE AMERICAN GERIATRICS SOCIETY 44:865-870 (1996)]. Use of estrogen for over one year reduced the risk of developing AD by 5% annually, but is of no demonstrable benefit for women who already have AD [BRITISH JOURNAL OF CLINICAL PHARMACOLOGY 52:647-653 (2001)]. Nonetheless, concerns over breast cancer & cardiovascular risks have dampened the interest in estrogen replacement therapy. Worse, because of increased vascular dementia, total dementias was actually observed to double in women using estrogen, according to one study [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION 289(20):2717-2719 (2003)]. Further studies have supported this conclusion [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION 291(24):2947-2958 (2004)]. There is debate, however, as to whether it was the medroxyprogesterone rather than the estrogen in the hormone replacement therapy studies that increased the risk of vascular events [CLINICS IN GERIATRIC MEDICINE; Hake,AM; 20(1):141-152 (2004)].
Soy protein has shown some of the benefits of estrogens, including reduced hyperphosphorylation of tau protein [BIOFACTORS; Kim,H; 12(1−4):243-250 (2000)]. Post-menopausal women not receiving hormone replacement therapy showed improved cognitive function with soya extract [PHARMACOLOGY, BIOCHEMISTRY AND BEHAVIOR; Duffy,R; 75(3):721-729 (2003)].
[For more about sex hormones and AD, see Sex Hormone Replacement in Older Adults]
Overweight women are particularly vulnerable to developing AD [ARCHIVES OF INTERNAL MEDICINE 163:1524-1528 (2003)]. (Which is to be distinguished from the weight loss that occurs due to deteriorating eating habits as AD progresses.) Whether or not this association is only due to elevated blood pressure, weight loss might be a promising way to reduce the risk of AD. Exercise (mental & physical) as well as measures used to prevent cardiovascular disease are of benefit in preventing AD [SCIENCE; Marx,J; 309:864-866 (2005)].
Transgenic AD mice subjected to chronic stress show enhanced Aß accumulation, which was apparently corticosteroid-related [JOURNAL OF NEUROSCIENCE; Carroll,JC; 31(40):14436-14449 (2011)]. (The glucocorticoid cascade hypothesis suggests that there is a positive feedback loop of rising cortisol levels in elderly humans — and many AD patients have substantially elevated blood cortisol.) A study of transgenic mice demonstrated that environmental enrichment exacerbated amyloid plaque formation [JOURNAL OF NEUROPATHOLOGY AND EXPERIMENTAL NEUROLOGY; Jankowsky,JL; 62(12):1220-1227 (2003)]. A subsequent study which claimed that the mice in the prior study had been subjected to excesssive stress, showed reduced Aß and increased levels of Aß-degrading enzyme neprilysin associated with environmental enrichment [CELL; Lazarov,O; 120(5):701-713 (2005)].
A mouse model of AD has shown reduced extracellular Aß with voluntary exercise [JOURNAL OF NEUROSCIENCE; Adland,PA; 25(17):4217-4221 (2005)]. A 14-year epidemiological study has shown a 29-50% reduction in AD incidence associated with physical activity [JOURNAL OF THE AMERICAN MEDICAL ASSOCIATION; Scarmeas,N; 302(6):627-637 (2009)] and a randomized study of over 4,000 men and women over age 65 showed reduced AD with physical activity [ARCHIVES OF NEUROLOGY; Laurin,D; 58(3):498-504 (2001)].
Forgetfulness & misplacement of items happen at all ages, but for the elderly these things are often rightly or wrongly interpreted as early signs of dementia. When reinforced by signs such as odd items in the refrigerator, leaving appliances on, repetitively asking the same question, peculiar dressing, inability to manage bills and increasing dependency upon others for everyday tasks, Alzheimer's Disease (AD) or another form of dementia may well be developing.
Many people find themselves somewhat reluctantly thrust into the role of caregiver for a spouse or other family member with AD. It can be especially difficult to see a loved-one "disappearing" before one's eyes. But while short-term memory may decay, long-term memories may be maintained along with special skills & talents. Those with a capacity for acceptance of the inevitable may be pleased to discover that dementia may allow their beloved to become more emotionally open — showing less inhibition to give & receive love. Hugs, massage & hand-holding (when slow & firm enough not to be alarming) can make bad situations much better.
Psychatric counseling is inappropriate for an AD patient. Arguing with an AD victim over lost money, eating habits, taking medications, inappropriate dressing or "illogical" behavior is fruitlessly upsetting and a waste of energy. Nonetheless, outbursts of anger are quickly forgiven because they are quickly forgotten by the person with AD.
Many times AD victims can help with housework or yardwork. Otherwise, they can be pleasantly occupied with colored pencils, clay, jigsaw puzzles, music, pets, gardening or exercise. Too much television can be debilitating and programs that are violent or noisy can be upsetting. Many areas have "day-care" centers for AD victims. If walking the streets can be done safely alone, it is a good idea for the AD victim to be given a Medic-Alert bracelet to wear — with name, address and phone number.
If an AD victim seems inclined to only eat cookies, ice cream & candy, indirect methods may be required to ensure that nutrition is adequate — along with an acceptance of the "junk-food" diet. Persons with AD may find bathing too complex. Less frequent washing and/or cleaning with a wet towel may be necessary adaptations. Small monetary allowances may be a means to deal with frequently lost money. It may be necessary to remove knobs from stoves and "safety-proof" a home with many of the techniques used for small children.
Although the prospect of a bedridden AD patient with no bowel or bladder control can be intensely worrying, many victims never reach that stage. Most bladder & bowel problems result from the AD victim not being reminded often-enough to go to the bathroom. Adult diapers and incontinence clothing can eliminate many problems in the later stages.
For patients who do reach the final stages, a great deal of devotion to the "former person" is required to deal with problems of urine, feces, feeding and bedsores from a "stranger" whose behavior can be erratic, irritable or dangerous. The majority of those in the vegetative state die from infections in lungs, urinary tract or bedsores associated with the increasing difficulty of maintaining cleanliness. Septicemia (microorganisms and their toxins in the bloodstream) can cause cardiac arrhythmia, clotting abnormalities and kidney or liver failure — leading to death. Caregivers may choose not to tube-feed and allow death by starvation. Death can be a relief when as the loved-one has already been long-lost.
Joining an Alzheimer's support group can help greatly in sharing care, sharing ideas and providing emotional reinforcement. The Alzheimer's Association and other national Alzheimer's organizations throughout the world can be good sources of information & help.
Alzheimer's Disease and other forms of dementia pose the most troubling dilemmas for cryonicists wishing to be cryopreserved upon legal death because of the risk that cryopreserving a demented brain may be of little value. As successful life-extensionists, cryonicists would be more likely than not to live long enough to develop AD. It is possible that a future technology could remove the amyloid plaques and neurofibrillary tangles from the brain of an Alzheimer's Disease victim and thereby restore mental function. Nonetheless, Alzheimer's Disease also involves definite neurodegeneration in which brain cells slowly die, possibly eventually leaving no self left worth preserving. There does, however, appear to be much redundancy in the brain, which allows stroke victims to recover memories and abilities seemingly destroyed. As with ischemic and ice-crystal damage, however, the most conservative approach is to prevent damage as much as possible rather than rely on a future technology that may be left with inadequate information to do a full restoration of the self to a condition of enduring youth & health.
Euthanasia is difficult enough to legalize for terminally ill patients. Alzheimer's Disease victims may not be deemed appropriate candidates for euthanasia even after their mental faculties have considerably deteriorated because although AD is inevitably fatal, AD is not a terminal disease (death of AD patients is usually due to infection). The situation is further complicated by the difficulty of making a definitive diagnosis, especially in the early stages of Alzheimer's Disease. Suicide would not prevent payment of life insurance to a cryonicist, but suicide is a legal mandate for autopsy — which typically means destruction of the brain because of long delays and because of the surgery. (For information on attempting to avoid autopsy, see Avoiding Autopsy for Cryonics.)
A large number of cryopreserved cryonicists have been AIDs victims. Jerry White, for example, contributed much of his time & money to cryonics before becoming a victim of AIDs. It was his intention to refuse food & water before he became too demented, but his fear of death and faultering judgment resulted in considerable mental decay before he was finally cryopreserved. An Alzheimer's Disease victim who still had the good judgment to refuse food & water for cryonics purposes would not be regarded as terminal. Such a death would probably be regarded as a suicide (leading to autopsy) and would risk legal action against any cryonicists who had the foreknowledge to attempt any form of cryonics rescue.
Other elderly people who have spent their lives working on behalf of cryonics organizations have allowed their cryonics coverage to lapse as they became increasingly demented. I have tried persuading such people, but to no avail. If they tell me they are no longer interested and don't keep their funding in place, what can I do? What are the true wishes of this person? Cryonics requires prognostications about the possibilities of future society and future technology — ideas not grasped by most people, much less those who are demented. The desire to continue or to not continue living is simpler, and is not necessarily related to cognitive ability.
Appointing another cryonicist as Durable Power of Attorney for Health Care may help, although in some cases this has been frustrated by a legal environment hostile to cryonics.
The problem of cryonicists with Alzheimer's Disease can only become worse if this disease is not cured. Aging cryonicists, increasing numbers of cryonicists and the success of technologies that can cure cardiovascular disease and cancer will increase the likelihood that people will survive only to become demented. The risk that the self will be destroyed before preservation is possible is a frightening one for cryonicists, which should make means of both preventing — as well as finding a cure for — Alzheimer's Disease a high priority.
![[GO TO BEN BEST'S HOME PAGE]](../homeback.gif) HOME PAGE
HOME PAGE